Мышечная дистрофия: виды и симптомы
Заболевание структур связочного аппарата – мышечная дистрофия – объединяет несколько форм патологии, некоторые из которых носят тяжелый характер и опасны для жизни.
Патология поражает глубокие слои мягких тканей, чаще всего относится к прогрессирующим формам. Это значит, что после обнаружения болезни, она будет постоянно развиваться, уменьшая силу мышечных тканей и диаметр волокон.
Прогрессирующая мышечная дистрофия неминуемо приводит к полному расщеплению части волокон, однако заболевание можно замедлить, предотвратив быструю дегенерацию тканей. При развитии болезни место мышц постепенно заменяет жировая прослойка.
Механизмы развития
Точных причин заболевания ученые обнаружить не могут, но выделяют мутации, отвечающие за развитие патологии. Так, в 100% случаев обнаруживается изменение в аутосомно-доминантном гене, который отвечает за выработку белка, участвующего в формировании и поддержании мускульных волокон.
На место развития мышечной дистрофии указывает поврежденная хромосома:
- если повреждена Х-хромосома, обнаруживают болезнь по типу Дюшена;
- если изменена 19 хромосома, появляется мотоническая форма болезни;
- недоразвитость мускульного скелета не связана с половыми хромосомами, обнаружить причины пока что не удается.
Провоцировать патологические изменения могут как наследственные, так и приобретенные формы мышечной дистрофии.
Первые признаки болезни
Вне зависимости от того, какой вид мышечной дистрофии обнаружен у пациента или начинает развиваться, все они сопровождаются общим набором признаков:
- начинается атрофия скелетных мышц;
- постепенно сокращается тонус мышечных тканей;
- изменяется походка, так как мышцы нижних конечностей неустанно слабеют;
- если такая патология обнаруживается у ребенка, он начинает постепенно терять мышечные навыки: падает голова, ему сложно сидеть и ходить;
- при этом боли в мышечных структурах не наблюдаются;
- сохраняется прежняя чувствительность;
- появляются частые падения;
- пациент жалуется на непроходящую усталость;
- разрастается соединительная ткань, вытесняя погибшие мускулы, что приводит к увеличению размеров мышц.


Мышечная дистрофия обрастает дополнительными симптомами по мере прогрессирования, каждый из них соответствует тому или иному типу патологии.
Виды заболевания и их признаки
Большая часть мышечных дистрофий относится к категории врожденных патологий, связанных с аномалиями в геноме. Однако есть некоторые формы, при которых мутации случаются в результате воздействия токсичных веществ.
Заболевание Дюшенна
Псевдогипертрофическая мышечная дистрофия Дюшена относится к прогрессирующему типу. Обнаруживают её еще в детском возрасте, так как симптомы носят выраженный характер и усиливаются быстро. Практически все пациент – мужского пола, но встречаются больные и среди девочек.
Симптомы мышечной дистрофии достаточно выражены уже в 2 года, до 5 лет они достигают своего пика:
- Патология начинается со слабости в тазовом поясе, затем страдают мускулы ног.
- Постепенно вовлекаются структуры всего скелета, что вызывает сбои со стороны внутренних органов.
- К 12-15 годам больной ребенок уже не может самостоятельно двигаться.

Эта прогрессирующая мышечная дистрофия в 100% случаев приводит к смерти до исполнения пациенту 30 лет. Большая часть не доживает до 20 лет.
Заболевание Штейнерта
Мышечная дистрофия Штейнерта развивается у взрослых от 20 до 40 лет и характеризуется поздними симптомами. В редких случаях патологию обнаруживают в младенчестве. По половой принадлежности особых корреляций врачи не отмечают. Протекает медленно, поддается сдерживанию.
Важно! Отличительная особенности нарушения – процесс проникает в структуры важных органов, провоцируя слабость лицевых мышц и других областей.
Волокно расщепляются медленно, но болезнь требует постоянного контроля со стороны врачей. Если мышечная дистрофия затронет легкие или сердце, может наступить быстрая смерть.
Заболевание Беккера
Синдром Беккера относится к прогрессирующим мышечным дистрофиям.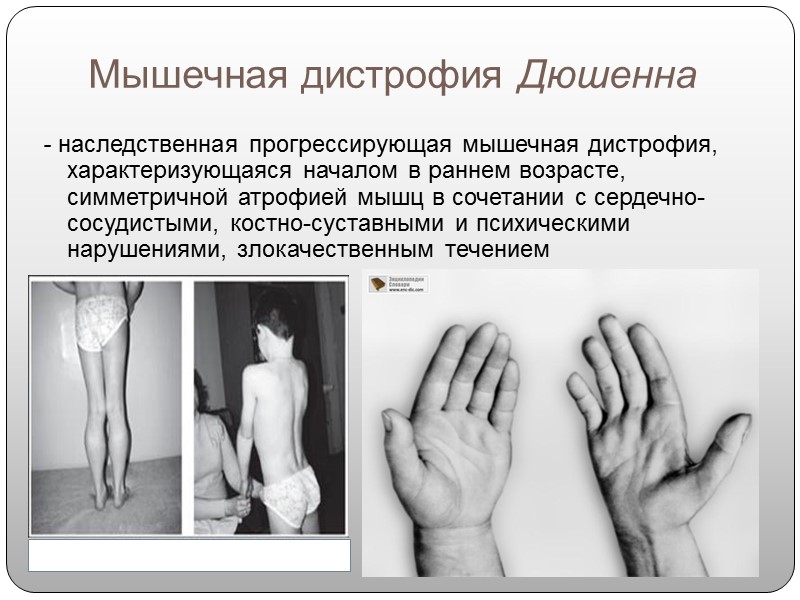 Встречается достаточно редко, развивается медленно. Чаще всего болезнь обнаруживают у людей низкого роста. Заболевание поддается терапии и легко контролируется, можно замедлить патологический процесс на 20-30 лет, сохранив стандартную работоспособность. Инвалидность наступает только в случае формирования дополнительных болезней или появления серьезных травм.
Встречается достаточно редко, развивается медленно. Чаще всего болезнь обнаруживают у людей низкого роста. Заболевание поддается терапии и легко контролируется, можно замедлить патологический процесс на 20-30 лет, сохранив стандартную работоспособность. Инвалидность наступает только в случае формирования дополнительных болезней или появления серьезных травм.
Юношеская дистрофия Эрба-Рота
Первые симптомы мышечная дистрофия провоцирует в период от 10 до 20 лет. Заболевание развивается медленно, начинается с пояса плеча и рук, затем в втягиваются другие мускулы. У человека прогрессирующие мышечные дистрофии вызывают сильное изменение осанки – грудная клетка отходит назад, а живот сильно выпирает вперед. Врачи описывают симптомы как «утиная походка».
Синдром Ландузи-Дежерина
Симптомы мышечной дистрофии впервые обнаруживаются у детей от 6 лет, но могут появляться в возрасте до 52 лет. Чаще всего первые признаки отмечают в период с 10 до 15 лет. Сначала поражаются лицевые мышцы, затем вовлекаются конечности и крупная мускулатура туловища.
Важно! Первый признак нарушения – неполное смыкание век во время сна. Затем перестают смыкаться губы, как во время отдыха, так и при бодрствовании, что сильно влияет на дикцию.
Развивается дистрофия мышц медленно, в течение длительного времени пациент сохраняет нормальную двигательную активность, может заниматься привычными делами. Атрофия тазового пояса, приводящая к инвалидизации, развивается в основном через 20-25 лет после обнаружения патологии. При адекватном лечении заболевание длительное время не проявляется сложными симптомами.
Алкогольная миопатия
Этот вид мышечной дистрофии никак не связан с геномом человека, так как развивается он только на фоне длительного употребления больших доз алкоголя. Сопровождается сильными болями в конечностях из-за распада мышечных волокон. Хроническая миопатия протекает с умеренными симптомами, а острая проявляется воспалением и приступами боли.
Дистальная форма
Дистальная мышечная дистрофия – это доброкачественное заболевание, которое сложно обнаружить из-за отсутствия выраженных симптомов. Нередко диагноз путают с невральной амиатрофией Мари-Шарко. Для дифференциального исследования необходима энцефалограмма головы. Общие признаки болезни сильно схожи с многими другими отклонениями.
Нередко диагноз путают с невральной амиатрофией Мари-Шарко. Для дифференциального исследования необходима энцефалограмма головы. Общие признаки болезни сильно схожи с многими другими отклонениями.
Миодистрофия Эмери-Дрейфуса
Для этой формы заболевания не существует специфических способов диагностики, она сильно похожа на синдром Дюшенна, однако есть специфические симптомы. Они проявляются достаточно редко, так как сам синдром возникает гораздо реже остальных видов дистрофии мышечного отдела.
В большинстве случаев патология развивается до 30 лет, страдают при этом мышцы сердца. Патология отличается наличием болезней сердца, однако проявляются они легкими симптомами. При отсутствии коррекции может приводить к смерти.
Нервно-мышечная форма
При этой форме мышечной дистрофии страдают нейронные связи, отвечающие за двигательную активность. В результате изменяются мышцы спинного мозга и более глубокие ткани. Нарушается строение ядра клеток нервов, первыми страдают лицевые мышцы, глаза.
У патологии много симптомов, часть из них затрагивает рецепторы чувств: ощущения могут усиливаться или уменьшаться. Иногда возникает головокружение, судороги, обнаруживаются болезни сердца и проблемы со зрением. Происходит сбой в работе потовых желез.
Конечностно-поясная дистрофия
Заболевание связано с наследственными нарушениями. Первыми в патологический процесс вовлекаются мышцы пояса и торса, за ними страдают верхние конечности. Лицевые мускулы практически никогда не втягиваются в заболевание. Прогрессирует состояние медленно, легко сдерживается медикаментами и не приводит к быстрой инвалидизации.
Окулофарингеальная форма болезни
Этому типу мышечной дистрофии свойственны поздние проявления, обнаруживается болезнь в зрелом возрасте. Чаще всего страдают определенные этнические группы. Симптоматика появляется в 25-30 лет:
- мышечная атрофия;
- птоз век и нарушение глотательной активности;
- невозможность движения глазных яблок.

Постепенно в процесс могут вовлекаться другие мышцы черепа, но это встречается не всегда. В некоторых случаях страдают мускулы плечевого пояса и шеи. Из-за этого возникают проблемы с речью и дикцией.
Заболевание и у детей
Прогрессирующая мышечная дистрофия у детей развивается по-разному и опасна больше осложнениями, чем первичной атрофией мускулов. Даже небольшая инфекция или респираторная патология может привести к летальному исходу из-за стремительного развития и вовлечения других органов. Заподозрить мышечную дистрофию иногда слишком сложно, родителям следует внимательно относиться к появлению симптомов:
- ребенок пытается подняться на носочки при ходьбе;
- обнаруживается задержка в развитии физическом и интеллектуальном;
- поражение мышечных структур начинается с позвоночника;
- сильно изменяется походка, становится развалистой;
- ребенок испытывает трудности во время бега, подъема по лестнице;
- позвоночник начинает деформироваться, из-за чего ребенок быстро утомляется;
- размер мышечных структур резко увеличивается из-за заполнения жировой прослойкой;
- увеличивается челюсть и промежутки между зубами;
- к 13 годам ребенок теряет способность нормально двигаться;
- развиваются сердечно-сосудистые заболевания.
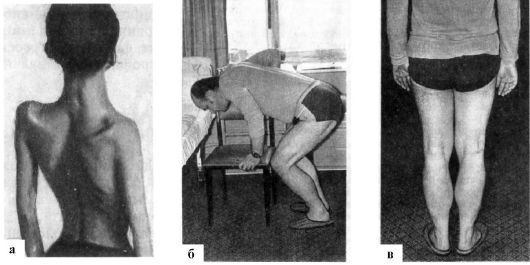
Формы заболевания могут иметь разные названия, но большая часть из них схожа симптомами.
Постановка диагноза
Диагностировать мышечную дистрофию можно после прохождения клинических обследований:
- Врач собирает симптомы и жалобы родителей или пациента.
- Затем исследуют ЭМГ.
- Берут нервно-мышечные волокна для проведения биопсии. Это один из самых точных и достоверных методов инструментального обследования.
- Затем изучают плазму крови на КФК, мышечная дистрофия сопровождается повышением показателей.
- Обязательно выделяют уровень креатинурии.
- Проводят МРТ мышечного слоя.
- Делают анализ крови на определение ферментов при подозрении на специфические болезни мышечной ткани.
- Проводят иммунологическое исследование у родителей, страдающих от заболевания, если они хотят завести ребенка.
При постановке диагноза обязательно указывают название белка, синтез которого недостаточен в организме.
Тактика терапии заболевания
Начинать лечение мышечной дистрофии нужно с устранения опасных симптомов, так как для решения генетических проблем актуальных методов коррекции пока что не существует. Например, при повреждении позвоночных мышц назначают препараты, улучшающие тонус.
Важно! Если патология опасна осложнениями со стороны сердечной системы, иногда имплантируют водитель сердечного ритма.
Большая часть препаратов относится к группе сильнодействующих средств и назначается врачом строго по рецепту. Кроме лекарств, врачи рекомендуют использовать ортопедические аппараты для укрепления мышц и кистей. Для укрепления мышечной ткани также используют анаболические стероиды.
Генная терапия – сложный и ненадежный метод лечения, однако она быстро развивается. Например, для лечения синдрома Дюшена используют искусственно созданный ген, который затем вживляют человеку. Для этого внутрь аденовируса помещают нужный ген и вводят в мышечную ткань.
Прогнозы и осложнения
Чаще всего мышечная дистрофия приводит к развитию опасных для жизни осложнений: нарушается работа легких и сердца, сокращается двигательная активность и наступает паралич, искривляется позвоночник, страдают интеллектуальные возможности.
Обнаружение мышечной дистрофии у пациента может стать приговором, но в отдаленной перспективе. Легче всего патологии протекают у взрослых. Если заболевание нашли у ребенка, шанс, что он проживет больше 20 лет, катастрофически мал. Однако поддерживающая терапия способна продлить активную жизнь пациента и минимизировать риск осложнений.
симптомы, механизм развития и прогноз
Мышечная дистрофия, или миопатия Дюшенна, – тяжелая наследственная патология, которая постоянно прогрессирует. Замедлить мышечное разрушение практически невозможно.
Связано это с врожденными изменениями. Впервые о миопатии Дюшенна заговорили в середине XIX века. Обнаружил эту патологию французский невролог. В тот момент был известен один тип течения болезни, через некоторое время выделили еще несколько способов развития состояния.
В тот момент был известен один тип течения болезни, через некоторое время выделили еще несколько способов развития состояния.
Этот тип болезни сильно похож на миодистрофию Беккера, но в то же время отличается от него сложностью и внешними признаками.
Миодистрофия Дюшенна обнаруживает у 1 ребенка из 4000. Этот тип патологии относится к самым распространенным мышечным дистрофиям, относится к врожденным заболеваниям.
Этиология нарушения
Одному из генов в структуре генома человека присвоили имя невролога, в честь которого и было названо отклонение. На мышечную дистрофию Дюшенна могут влиять разные факторы:
- кровосмешение;
- предрасположенность генетического характера, например, при наличии миопатии Дюшенна у кого-либо из родни;
- неправильный синтез мышечных волокон, ускоренное распространение и замещение жировой прослойкой, соединительными волокнами;
- наследственные формы синдрома Дюшенна, чаще всего переходящие от матери;
- мутация генома при формировании во время беременности;
- аномалии в хромосомных структурах неясного происхождения;
- сильные нарушения в развитии дистрофина;
- патологические изменения биохимии в крови.
Также миопатия Дюшенна формируется при заболеваниях соединительной ткани, не связанных напрямую с генетическими отклонениями.
Характеристика наследственной патологии
Генетическая природа заболевания была сразу же доказана после обнаружения синдрома в 1868 году. Эта патология почти идентична с миодистрофией Беккера, то есть, обладает теми же генетическими предпосылками для формирования.
Однако миодистрофия Беккера отличается иными симптомами. Для болезни характерны следующие особенности:
- диагностируется у мальчиков до 5 лет;
- прогрессирует стремительно;
- у девочек никогда не обнаруживается;
- атрофия мышц обладает ступенчатым развитием – сначала страдает тазовый пояс;
- затем вовлекаются мышцы ног;
- после этого миопатия Дюшенна поражает мышцы спины, плеч;
- завершается прогрессирующая мышечная дистрофия Дюшенна поражением рук;
- специфический признак нарушения – деформация позвоночника, чаще встречающаяся в форме кифоза или лордоза;
- миодистрофия Дюшенна почти всегда сопровождается повреждениями грудины и стоп, они становятся неправильной формы, сильно меняют тело человека;
- при патологии, в отличие от миодистрофии Беккера, появляется повреждение левого сердечного желудочка, аритмия и кардиопатия;
- примерно у 30% пациентов развивается олигофрения.

Мышечная дистрофия Дюшенна никогда не протекает в легкой степени, всегда имеет крайне неблагоприятный прогноз. Развивается быстро, возможность ходить пациент утрачивает уже к 12 годам. При мышечной дистрофии Дюшенна смерть наступает из-за инфекции бронхов или легких, после остановки сердца.
Симптомы нарушения
Первые признаки миопатии Дюшенна встречаются уже в возрасте 1,5 лет. В редких случаях их не удается заметить до 5 лет. Проявляются признаки заболевания Дюшенна сначала в легкой степени. Их комбинация зависит от общего состояния здоровья:
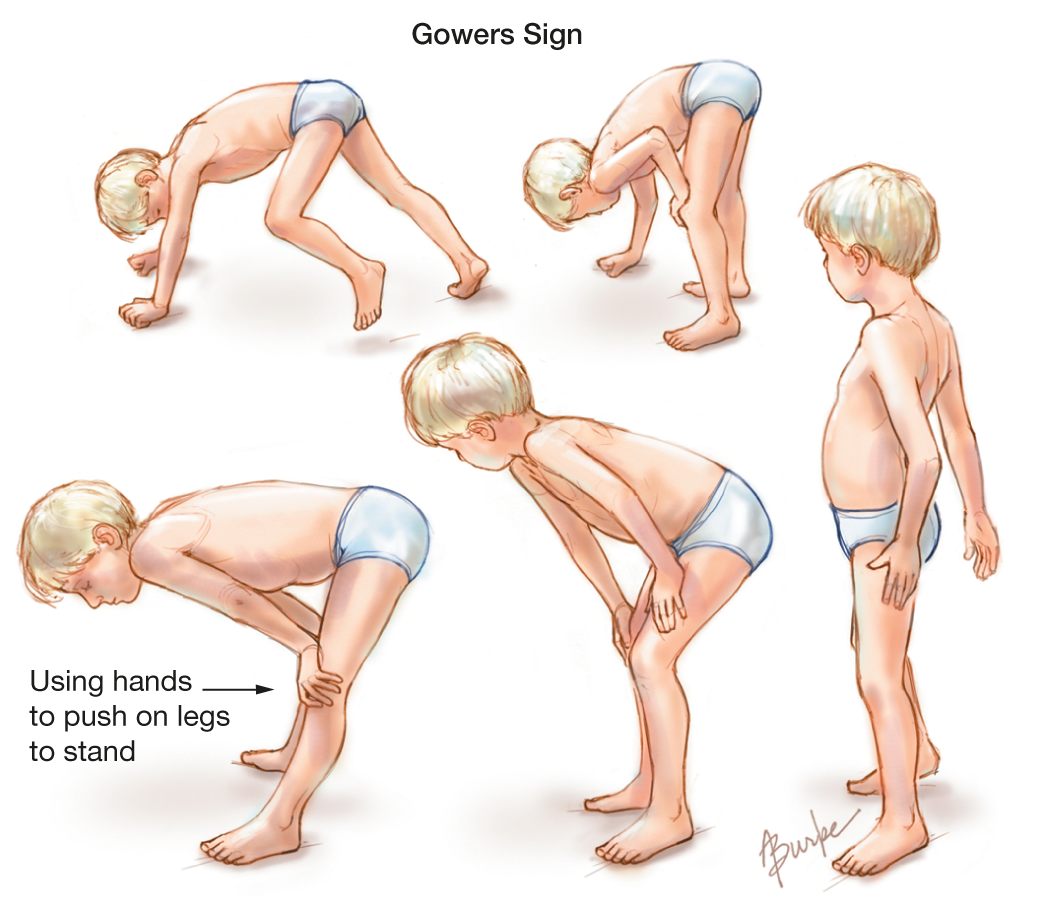
- у ребенка возникает сильная неустойчивость, наблюдается неловкость в движении, он часто падает и очень медлителен;
- миопатия Дюшенна сопровождается тем, что во время ходьбы ребенок вихляет, постоянно спотыкается, в результате чего малыш боится вставать на ноги, возникает выраженная двигательная пассивность;
- со временем при мышечной дистрофии Дюшенна становится видна «утиная» походка с выпяченной вперед грудью и отведенными назад лопатками;
- если ребенок сидит или лежит, то принять стоячее положение при мышечной дистрофии Дюшенна становится сложно;
- при попытках принять стоячее положение, малыш словно встает на лесенку, поднимается вверх задом;
- возникает гипертрофия мышц, они заполняются жировой тканью;
- также миодистрофия Дюшенна захватывает работу сердца, в результате чего развиваются патологии и недостаточность;
- мышечная дистрофия Дюшенна часто сопровождается и другим признаком – появляются нарушения в биоптатах скелета;
- постепенно изменяется положение крупных суставов, начинается деформация стоп;
- миопатия Дюшенна в 100% случаев приводит к полной инвалидности пациента, ему требуется кресло;
- в 15 лет при мышечной дистрофии Дюшенна наступает глубокая инвалидизация, опасная остановкой сердца и хронические или постоянно рецидивирующие нарушения в легких.

На фоне мышечной дистрофии Дюшенна у маленького пациента развивается острая депрессия, которую дети с трудом переносят. Нередко причиной смертности при миодистрофии Беккера и Дюшенна становится суицид.
Диагностика заболевания
Мышечная дистрофия Дюшенна крайне тяжело поддается диагностики. Для этого привлекают комплекс методов. Первое, что нужно пройти при подозрении на миопатию Дюшенна, – это ЭКГ. Для подтверждения диагноза необходимо, чтобы анализ показал нарушения стенки левого желудочка.
Следующий этап – это определение уровня дистрофина, который не меняется в сторону обычной дистрофии. Также необходимо сдать кровь на биохимический анализ. Если есть миодистрофия Беккера или болезнь, названная в честь французского невролога, отмечается высокий уровень КФК.
Дополнительно нужно пройти ЭМГ, генодиагностику, а также биопсию мышц. Именно последний анализ позволяет установить болезнь с достаточно высокой точностью. Электромиография не уступает в эффективности с точки зрения постановки диагноза «мышечная дистрофия Дюшенна».
Тактика лечения заболевания
Чтобы лечение мышечной дистрофии Дюшенна было эффективным, нужно четко следовать намеченному плану после постановки диагноза. Излечению болезнь никогда не поддается полностью, но можно значительно облегчить жизнь пациента. Современная медицина способна замедлить миопатию Дюшенна следующими методами:
- Тактика при обнаружении болезни до 5 лет. Радикального лечения миодистрофии Дюшенна не требуется. Нужна генетическая консультация и постоянна поддержка родителей больного ребенка.
- Лечение миопатии Дюшенна до 8 лет. В этом случае нужна поддержка мышечных тканей. Врачи назначают глюкокортикостероиды для замедления прогресса болезни: «Преднизолон» или «Дефлазакорт».
- Терапия от 8 до 20 лет. В этом случае значительно ослабляются мышцы, мышечная дистрофия Дюшенна приводит ребенка к инвалидному креслу.
- Терапия от 20 лет. В этом случае препараты частично перестают действовать, прогрессируют заболевания дыхательных путей.
Миопатия Дюшенна требует постоянного приема некоторых групп витаминов (B, E), а также кальция, гормонов анаболиков, калия и некоторые виды аминокислот. Обязательно при мышечной дистрофии Дюшенна назначают инъекции АТФ, «Ретаболила», глютаминовой кислоты.
Важно! Поддержать здоровье при мышечной дистрофии Дюшенна можно и другими методами – ЛФК и электрофорезом.
ЛФК проходят небольшими курсами с обязательным участием терапевта. Также врачи рекомендуют делать массаж. Для электрофореза при миопатии Дюшеннанеобходимо использовать такие вещества, как липаза, хлорид кальция, «Прозерин».
В тяжелых случаях всё лечение проводят в домашних условиях, если есть медицинские возможности для организации сложной терапии специальными приборами.
Обязательное условие для лечения миопатии Дюшенна – постоянное наблюдение у кардиолога. Также необходимо составить грамотное меню. При заболевании нужно есть много овощей, приготовленных на пару, фруктов, растительных жиров и нежирного мяса.
 Запрещено употребление алкоголя, кофеина и крепкого чая.
Запрещено употребление алкоголя, кофеина и крепкого чая.Последствия и осложнения
В 100% случаев миопатия Дюшенна сопровождается тяжелыми последствиями для организма и сильно укорачивает жизнь. Пациент всегда умирает от осложнений заболевания – остановки сердца или легочной инфекции.
Если мышечную дистрофию Дюшенна удалось обнаружить в раннем возрасте, есть шанс, что человек доживет до 30 лет. Но только при условии адекватной терапии и комплексного подхода. Среди осложнений миопатии Дюшена нередко выделяют остеопороз, поражения позвоночника и суставов, а также патологии пищеварительной системы.
Мышечная дистрофия Дюшенна – тяжелое генетическое расстройство, терапия которого не способна оградить человека от одного исхода – смерти. В некоторых случаях пациентам удается прожить больше 20 лет после постановки диагноза. В других случаях младенцы умирают в течение первого года жизни.
Прогрессирующая мышечная дистрофия — Знаешь как
От греческого myos —мышца, pathos — страдание) — хроническое наследственное заболевание, проявляющееся симметричными атрофиями и слабостью мышц. Встречается как в семьях (наследуется по аутосомно-доминантному, по аутосомно-рецессивному и по рецессивному сцепленному с полом типам), так и в виде спорадических случаев, причем по клиническому течению между ними не обнаруживается каких-либо различий. Толчком к проявлению болезни могут служить травмы, инфекции, интоксикации, физическое переутомление, истощение.
Встречается как в семьях (наследуется по аутосомно-доминантному, по аутосомно-рецессивному и по рецессивному сцепленному с полом типам), так и в виде спорадических случаев, причем по клиническому течению между ними не обнаруживается каких-либо различий. Толчком к проявлению болезни могут служить травмы, инфекции, интоксикации, физическое переутомление, истощение.
Патоморфологические изменения сводятся в основном к перерождению мышечных волокон (атрофии и гипертрофии) и постепенной замене их соединительной и жировой тканью. Мышечные волокна истончены и неравномерны, местами отмечаются продольное расщепление и образование в них вакуолей, очаги некроза в саркоплазме.
При гистохимическом исследовании в мьшщах определяется увеличение кислых мукополисахаридов. Изменения нервной системы выражены незначительно и непостоянно. Имеются немногочисленные данные о наличии дистрофических изменений в вегетативных ядрах гипоталамуса, ствола и спинного мозга, об уменьшении количества клеток передних рогов, вздутиях и истончении осевых цилиндров, исчезновении фибриллярной структуры в моторных бляшках и т. д.
д.
В основе патогенеза болезни лежат расстройства обмена веществ в мышечной ткани вследствие ферментного блока, что приводит к нарушению синтеза и ускоренному распаду мышечного белка. Наиболее убедительны представления о дефекте обмена в самом мышечном волокне. Меньше сторонников имеет теория расстройств центральных механизмов вегетативно-эндокринной регуляции трофики мышц, переднероговых или сосудистых нарушений. Возможно предположение о распространении на нервную ткань эффекта мутантного гена или о существовании разных по патогенезу форм миопатии (генокопий).
Клиническая картина характеризуется постепенно нарастающими симметричными атрофиями и слабостью мышц верхних и нижних конечностей, лица, шеи, туловища. Поражение мышц конечностей и туловища приводит к затруднению передвижения прогрессивная мышечная («крыловидные лопатки»), выпячиванию живота («лягушачий живот»), к сужению талии больного («осиная талия»).
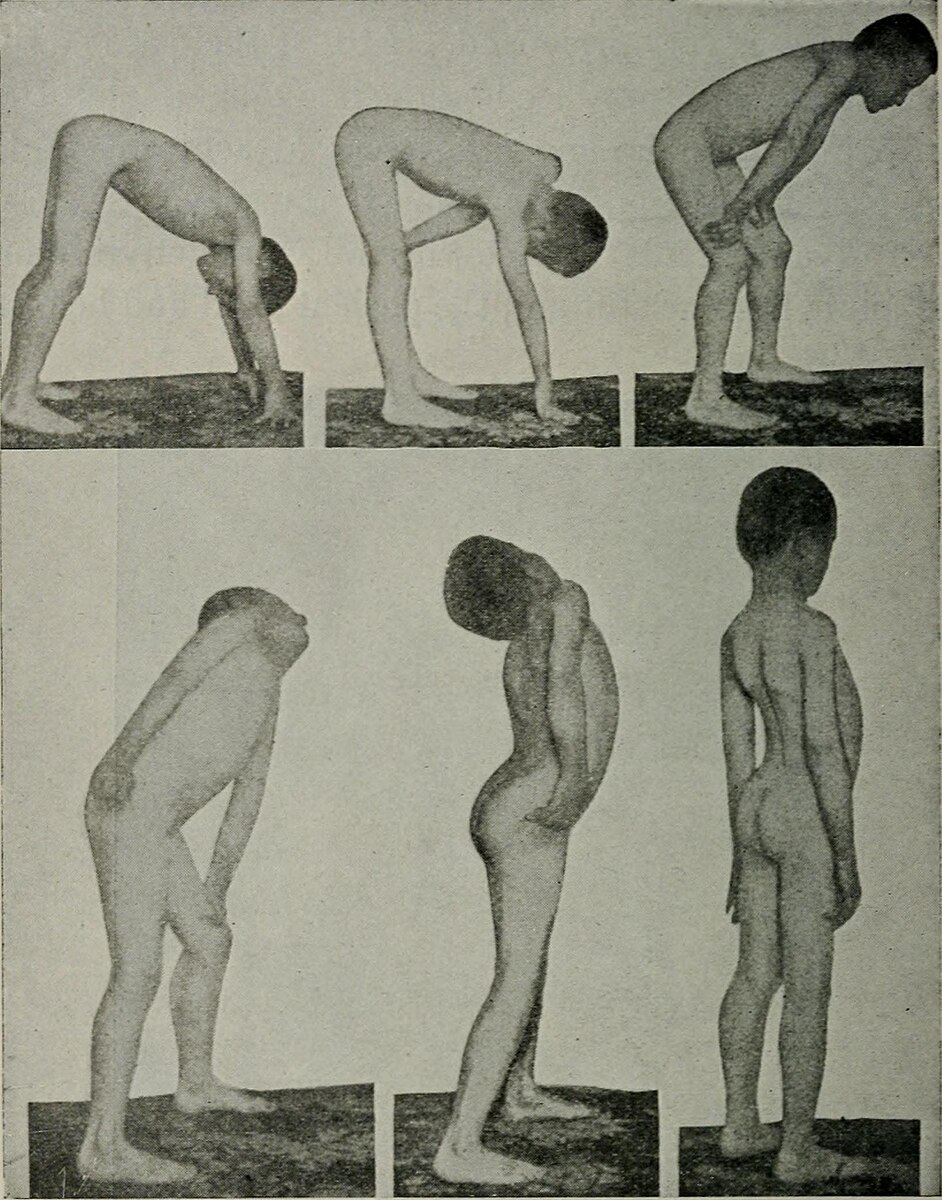
Вследствие распространения процесса на лицевые мышцы больной не может наморщить лоб, плотно закрыть глаза («миопатическое лицо»).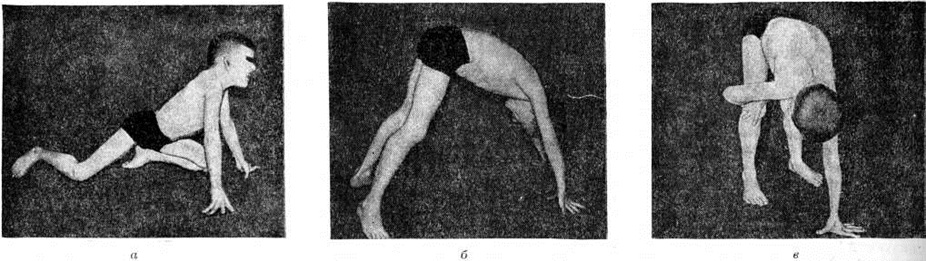 При смехе и улыбке углы рта чрезмерно растягиваются («поперечная улыбка»). Больные не могут встать со стула без помощи рук; для того чтобы подняться с пола, они вынуждены встать сначала на четвереньки, затем выпрямить ноги и постепенно, поочередно упираясь руками в голени, колени и бедра, поднять туловище.
При смехе и улыбке углы рта чрезмерно растягиваются («поперечная улыбка»). Больные не могут встать со стула без помощи рук; для того чтобы подняться с пола, они вынуждены встать сначала на четвереньки, затем выпрямить ноги и постепенно, поочередно упираясь руками в голени, колени и бедра, поднять туловище.
Наряду с атрофичными мышцами могут наблюдаться псевдогипертрофии некоторых мышц вследствие отложения жира и разрастания соединительной ткани. Особенно часто увеличиваются в размерах икроножные мышцы. Избирательность поражения (проксимальные отделы, например, страдают больше дистальных) постепенно приводит к образованию деформации и контрактур в тазобедренных, коленных, голеностопных суставах, в пальцах рук. Выделен особый миосклеротический вариант болезни (Сестана — Лежонна), при котором ретракции мышц доминируют в клинической картине.
Симптомов органического поражения нервной системы не бывает. Наблюдаемое снижение, а затем и исчезновение сухожильных рефлексов обусловлено мышечными атрофиями. Чувствительность, координация движений, тазовые функции сохранены. Имеют место разнообразные вегетативные нарушения — цианоз кистей и стоп, повышенная потливость и похолодание конечностей, асимметрии кожной температуры и др. Нередко вследствие вовлечения гладких мышц отмечаются изменения внутренних органов: расширение границ и глухость тонов сердца, затруднение внешнего дыхания, вялая перистальтика кишечника и др.
Чувствительность, координация движений, тазовые функции сохранены. Имеют место разнообразные вегетативные нарушения — цианоз кистей и стоп, повышенная потливость и похолодание конечностей, асимметрии кожной температуры и др. Нередко вследствие вовлечения гладких мышц отмечаются изменения внутренних органов: расширение границ и глухость тонов сердца, затруднение внешнего дыхания, вялая перистальтика кишечника и др.
Самый частый вариант прогрессирующей мышечной дистрофии — ювенильная (поясноконечностная) форма Эрба — Рота, которая наследуется по аутосомно-рецессивному и реже аутосомно-доминантному типам (в последнем случае пенетрантность относительно небольшая). Первые симптомы появляются в детском и юношеском возрасте. Атрофический процесс начинается с мышц тазового пояса и проксимальных отделов ног или реже с плечевого пояса и постепенно распространяется на другие мышцы туловища и конечностей. Течение болезни медленнопрогрессирующее.
Псевдогипертрофическая форма Дюшенн а —одна из относительно частых форм миопатии. Передается рецессивно, сцепленно с полом (с Х-хромосомой, поэтому болеют мальчики). Пенетрантность патологического гена высокая. Начинается болезнь в детском возрасте (в первые 5 лет жизни) со слабости мышц тазового пояса и ног, затем вовлекаются проксимальные отделы рук и другие мышцы. Характерны псевдогипертрофии икроножных, а иногда ягодичных, дельтовидных и других мышц. Рано выпадают коленные рефлексы. Часты вегетативно-эндокринные расстройства (ожирение, гипергидроз и др.). В отличие от других форм миопатии отмечается более заметное снижение интеллекта. Течение заболевания быстропрогрессирующее — дети к 10—12 годам с трудом ходят, а к 13— 15 — становятся практически полностью обездвиженными.
Передается рецессивно, сцепленно с полом (с Х-хромосомой, поэтому болеют мальчики). Пенетрантность патологического гена высокая. Начинается болезнь в детском возрасте (в первые 5 лет жизни) со слабости мышц тазового пояса и ног, затем вовлекаются проксимальные отделы рук и другие мышцы. Характерны псевдогипертрофии икроножных, а иногда ягодичных, дельтовидных и других мышц. Рано выпадают коленные рефлексы. Часты вегетативно-эндокринные расстройства (ожирение, гипергидроз и др.). В отличие от других форм миопатии отмечается более заметное снижение интеллекта. Течение заболевания быстропрогрессирующее — дети к 10—12 годам с трудом ходят, а к 13— 15 — становятся практически полностью обездвиженными.
Плечелопаточно-лицевая форма Ландузи — Дежерина наследуется по доминантному типу с полной пенетрантностью патологического гена. Начинается в возрасте 20—25 лет, отличается значительной атрофией лицевых мышц, особенно круговых мышц глаза и рта. Развивается гипомимия, невозможность зажмурить глаза. В дальнейшем атрофии захватывают мышцы плечевого пояса, туловища, проксимальных отделов рук и дистальных отделов ног. Иногда наблюдаются умеренные псевдогипертрофии отдельных мышц. Сухожильные рефлексы долго остаются сохранными. Заболевание течет сравнительно доброкачественно.
В дальнейшем атрофии захватывают мышцы плечевого пояса, туловища, проксимальных отделов рук и дистальных отделов ног. Иногда наблюдаются умеренные псевдогипертрофии отдельных мышц. Сухожильные рефлексы долго остаются сохранными. Заболевание течет сравнительно доброкачественно.
Офтальмоплегическая миопатия (окулярная миопатия) передается по аутосомно-доминантному типу, но встречается много спорадических случаев, что свидетельствует о невысокой пенетрантности патологического гена. Заболевают чаще в зрелом возрасте. Процесс захватывает в основном наружные мышцы глаз: развивается двусторонний птоз, затруднение при движении глаз в стороны и вниз. Поскольку процесс симметричен, двоение отмечается редко. Внутренние мышцы глаз обычно не страдают. Постепенно заболевание генерализуется — появляется слабость другой лицевой мускулатуры, мышц гортани и глотки, плечевого пояса и т. д.
Лопаточно-перонеальная форма Давиденкова передается по доминантному типу, характеризуется поздним началом (в 25—30 лет), преимущественным вовлечением мышц проксимальных отделов верхних конечностей и плечевого пояса и дистальных отделов ног.
Дистальная миопатия наследуется по доминантному типу. Начинается в возрасте 20—25 лет. Ее отличает преимущественное поражение мышц предплечий, кистей, голеней, стоп, медленное прогрессирование.
Статья на тему Прогрессирующая мышечная дистрофия
Гипотезы патогенеза прогрессирующих мышечных дистрофий
23.08.2011г.
Механизмы развития прогрессирующих мышечные дистрофий остаются загадкой и имеется несколько конкурирующих теорий их происхождения, каждая из которых отдает предпочтение определенному патогенетическому механизму их формирования.
Несомненно, что большинство мышечных дистрофий генетически обусловлено, однако механизм реализации генетически запрограммированных нарушений неясен. Остановимся на нейротрофической теории, гипотезе мышечной гипоксии, гипотезе «дефектных мембран» и роли нарушений системы цАМФ в формировании миодистрофического процесса.
Нейротрофические теории происхождения мышечных дистрофий
Вопрос о нарушении нервных влияний в формировании мышечных дистрофий активно обсуждается в литературе со времени первых описаний этого заболевания (Dushen, 1848; Erb, 1848).
В дальнейшем неоднократно появлялись описания больных и посмертно верифицированных случаев, когда при прогрессирующих мышечных дистрофиях выявились структурные изменения головного мозга, клеток передних и боковых рогов спинного мозга, внутримышечных нервов и их терминалей, а также сочетания классических форм миодистрофий с патологией подкорковых и гипоталамических отделов мозга. Однако механизмы этих влияний оставались неясными, и большинство неврологов скептически относились к значимости этих находок.
Другое направление исследований базируется на развивающихся в последние годы представлениях о нервной трофике как одной из форм межклеточных взаимоотношений, а именно о влиянии мотонейрона на мышечное волокно.
Как известно, с внедрением в клиническую неврологию морфологических и электрофизиологических методов исследования прогрессирующие мышечные дистрофии были подразделены на две группы: первично-мышечные дистрофии и вторичные неврогенные атрофии.
Согласно классическим представлениям основанием для отнесения заболевания к неврогенным явилось наличие «пучковой атрофии» мышечных волокон и наличие электромиографических признаков денерваций
— потенциалов фасцикуляций, фибрилляций и положительных острых волн, а также потенциалов действия, увеличенных вследствие реиннервации двигательных единиц в пораженных мышцах [Рот В. К., 1891; Даркшевич Л. О., 1904; Kugelberg, 1948; Buchthal et al., 1957].
К., 1891; Даркшевич Л. О., 1904; Kugelberg, 1948; Buchthal et al., 1957]. В течение последующих лет в литературе появилось большое число сообщений, позволивших подвергнуть пересмотру специфичность морфологических и электрофизиологических критериев первично-мышечной и «неврогенных» форм патологии. Показано, что на ранних стадиях денервационного процесса наблюдаются атрофии отдельных мышечных волокон, равномерно распределенных по всему поперечнику мышц.
При некоторых формах патологии мотонейрона, например при спинальной амиотрофии Кугельберга — Веландер и при хирургических перерезках некоторых нервных стволов в эксперименте, может в течение длительного времени отмечаться «рассыпная атрофия» отдельных мышечных волокон, и только при специальных методах гистохимического обследования мышц удается выявить группировку мышечных волокон. Отмечено также изолированное поражение мышечных волокон 1-го или 2-го типа при несомненно денервационных амиотрофиях.
Распространенное мнение о том, что любая денервация обязательно ведет к разрастанию концевых ветвлений двигательных нервов, созданию эффективных контактов этих волокон с мышечными волокнами и увеличению площади, занимаемой ДЕ, следует считать не полностью соответствующим действительности.
При некоторых интоксикациях у экспериментальных животных, ряде воздействий у человека и некоторых синдромах, наблюдавшихся в клинике, в течение длительного времени могут обнаруживаться денервационные изменения без явлений реиннервации по результатам клинического, морфологического и электрофизиологического исследования мышц (Гехт Б. М. и др., 1980; Engel, 1973].
Наблюдение за структурой мышечных волокон в зонах, где, по данным электромиографического обследования, можно предположить наличие укрупненных двигательных единиц на фоне полей однотипных мышечных волокон, выявляет двух- и трехзонные «волокна — мишени», типичные для некоторых генетически обусловленных форм мышечной патологии [Engel, 1973].
Приведенные наблюдения свидетельствуют о неспецифичности морфологических и электрофизиологических данных и недостаточности их для решения вопроса о неврогенном или миогенном характере мышечных дистрофий. Кроме того, результаты упомянутых исследований позволяют предположить возможность утрачивать мотонейроном не только все свои функции целиком, как это имеет место при разрушении мотонейрона или его аксона, но и частично.
По мнению W. Engel (1973), мотонейрон может выключаться полностью в результате его гибели или разрушения аксона (in toto) либо частично (in portio). В последнем случае вначале будут выключаться отдельные его волокна, в первом — все элементы двигательной единицы. Возможно выключение мотонейронов 1-го или 2-го типа или комбинация этих поражений. В этих случаях будет наблюдаться нарушение мышечных волокон соответствующего типа или мышечных волокон обоих типов.
Второе положение Engel заключается в допущении наличия многих влияний нерва на мышцу. Так, Engel допускает наличие трофического фактора, обеспечивающего созревание мышечных волокон; фактора, поддерживающего нормальное состояние мышечных волокон, сохранение его типа обмена и структуры; фактора, поддерживающего нормальную чувствительность мышечных волокон к ацетилхолину; фактора, препятствующего фибрилляции мышечных волокон; фактора, обусловливающего формирование постсинаптических структур и др.
Недостаточность каждого из трофических факторов или их комбинации, по мнению автора, приводит к определенной форме патологии мышц. Если к этому прибавить возможность изолированного поражения мотонейронов 1-го или 2-го типа, а также возможность поражения мотонейронов целиком или частично, то число вариантов патологии мышц, которые могут возникнуть при нарушении нервно-трофических влияний, будет соответствовать существующему многообразию форм патологии периферического нейромоторного аппарата.
Рассматривая гипотезу Engel, большинство исследователей отмечают ее стройность, четкое соответствие с имеющимися в литературе нейрофизиологическими данными, а также то обстоятельство, что эта гипотеза способна объяснить ряд нейрофизиологических феноменов, ранее неясных, например, возможность появления миотонических и псевдомиотонических феноменов и миопатических изменений ЭМГ на определенных стадиях развития неврогенных амиотрофий.
Другая гипотеза, подчеркивающая роль нарушений нервной трофики в происхождении мышечных дистрофий, сформулирована Mac Comas (1971). Суть ее состоит в том, что при развитии патологического процесса, обусловленного поражением мотонейронов, помимо крайних состояний — нормальное состояние клетки и ее гибель, допускается наличие нескольких переходных состояний — состояния «больного мотонейрона».
На ранних стадиях нарушений его функции наблюдается нарушение нервно-мышечной передачи в отдельных концевых ветвлениях аксона. По мере развития болезни число пораженных терминалей все больше увеличивается. На следующей стадии наиболее пораженные мышечные волокна подвергаются обратимым, а затем и необратимым денервационным изменениям. Затем погибают терминальные ветвления мотонейрона, его аксон и в конечной стадии процесса — сам мотонейрон.
В качестве одной из форм нарушений функций мотонейрона предполагается утрата его способности реиннервировать рядом лежащие денервированные мышечные волокна. Автор допускает и «хронические болезни мотонейрона» — состояния, при которых мотонейрон в течение длительного времени находится на определенной стадии нарушения его функций.
Следует указать, что одно из основных положений Mac Comas о возможности обратимой дисфункции мотонейронов подтверждено в экспериментальных условиях и при наблюдении за больными, получавшими с лечебной целью винкристин или винбластин [Covanagh Р., 1964]. Хуже обстоит дело с другим положением автора, лежащим в основе данной теории, — предположении об уменьшении числа активно функционирующих мотонейронов при ряде форм мышечных дистрофий [Mac Comas et al., 1974].
Большинство исследователей, повторивших исследование Mac Comas, подтвердили возможность применения его метода при изучении хронических нейрональных заболеваний, однако не смогли подтвердить основного положения Mac Comas об уменьшении числа двигательных единиц при миодистрофиях [Ballantyne, Hansen S., 1974].
В заключение необходимо указать, что признание нейротрофической гипотезы мышечных дистрофий не означает умаления роли наследственного фактора в их происхождении.
Вопрос ставится иным образом: являются ли мышечные дистрофии результатом наличия нарушений обмена непосредственно в мышечных волокнах либо первичный генетически обусловленный дефект локализован в мотонейронах, а изменения мышечных волокон являются вторичными, обусловленными нарушениями трофических влияний нерва на мышцу.
«Нервно-мышечные болезни»,
Б.М.Гехт, Н.А.Ильина
Читайте далее:
Дистрофии мышечные прогрессирующие — это… Что такое Дистрофии мышечные прогрессирующие?
группа наследственно обусловленных нервно-мышечных заболеваний, характеризующихся прогрессирующей мышечной слабостью, атрофией мышц, двигательными нарушениями. Различные формы Д. м. п. отличаются разным типом наследования, вариабельностью возраста начала заболевания, преимущественной локализацией поражения мышц и другими признаками.
К наиболее часто встречающимся формам относится псевдогипертрофическая (Х-хромосомная) злокачественная миодистрофия Дюшенна; псевдогипертрофическая (Х-хромосомная), доброкачественная миодистрофия Беккера — Кинера; поясно-конечностная, юношеская (аутосомно-рецессивная) миодистрофия Эрба — Рота; плече-лопаточно-лицевая (аутосомно-доминантная) миодистрофия Ландузи — Дежерина. Редкими являются дистальная, офтальмоплегическая, бульбарная и другие смешанные и переходные формы. Существует несколько гипотез патогенеза Д. м. п. Наиболее достоверной считают теорию дефектных мембран, согласно которой в основе патогенеза Д. м. п. лежит нарушение структуры сарколеммы и саркоплазматической сети. В связи с повышенной проницаемостью сарколеммы происходит «утечка» в кровь различных веществ из мышцы (ферментов, аминокислот, углеводов, креатина и др.). Это позволяет использовать биохимические методы с целью диагностики отдельных форм, определения тяжести, прогноза заболевания, оценки эффективности терапии, а также для выявления гетерозиготного носителя. Клиническая картина. Характерными симптомами Д. м. п. являются мышечная слабость и атрофия мышц, которые могут проявляться в различные возрастные периоды, но чаще развиваются в детском и юношеском возрасте. Дети поздно начинают ходить, быстро утомляются, неуклюжи в ходьбе, спотыкаются при беге, часто падают, с трудом поднимаются по лестнице. Двигательные нарушения постепенно прогрессируют. Возникает миопатическая утиная походка. В случае поражения мышц тазового пояса и конечностей затруднен переход из горизонтального положения в вертикальное; при поражении дистальных групп мышц ног появляется петушиная походка. Стойкость и нарастание двигательных нарушений позволяют диагностировать миодистрофию уже на ранних стадиях заболеваниях. При обследовании больного обнаруживают генерализованную или локальную атрофию мышц. Локальная атрофия мышц выявляется лишь на ранних стадиях заболевания, по мере прогрессирования патологического процесса атрофия мышц приобретает генерализованный характер вплоть до мышечной кахексии. Атрофированные мышцы истончены, дряблые при пальпации, однако следует отметить, что наряду с атрофией мышц выявляется псевдогипертрофия (замещение атрофированных мышц жировой клетчаткой, соединительной тканью). Миодистрофический процесс сопровождается поражением соединительной ткани, миосклерозом, развитием сухожильно-связочных ретракций, ограничением объема движений в суставах, укорочением пяточного (ахиллова) сухожилия, контрактурами. Одновременно с развитием мышечных атрофий снижаются сухожильные рефлексы, в первую очередь коленные.Миодистрофия Роттауфа — Мортье начинается в возрасте 8—9 лет, отличается выраженным миосклерозом, развитием сухожильных ретракции и контрактур в локтевых, голеностопных суставах, ригидности позвоночника. Мышечные атрофии преобладают в лопаточно-плечевой области и в дистальных отделах конечностей. Псевдогипертрофии отсутствуют, интеллект сохранен. Течение заболевания медленное, больные длительно сохраняют подвижность и обслуживают себя.
Поясно-конечностная юношеская миодистрофия Эрба — Рота. Тип наследования аутосомно-рецессивный, имеются спорадические случаи. Заболевание проявляется в возрасте 13—16 лет, однако первые его признаки могут наблюдаться в раннем детском возрасте (дистрофия Лейдена). Характерными симптомами являются слабость и атрофия мышц тазового пояса и бедер, мышц живота и туловища, что проявляется гиперлордозом позвоночника, выпячиванием живота, утиной походкой, затруднением при переходе из горизонтального положения в вертикальное. Генерализация атрофий происходит по восходящему типу. Псевдогипертрофии икроножных мышц, ретракции и контрактуры выражены умеренно, интеллект сохранен. Кардиомиопатия проявляется в поздних стадиях заболевания. Атрофия дыхательных мышц, деформация грудной клетки и позвоночника приводят к нарушению функции внешнего дыхания, легочно-сердечной недостаточности. Смерть больных обычно наступает от бронхопневмонии. С помощью электромиографии выявляют снижение амплитуды, полифазность биопотенциалов. При биохимическом исследовании обнаруживают гиперферментурию, гипер-миноацидурию, креатинурию, пентодурию и др., но выраженные в меньшей степени, чем у больных миодистрофией Дюшенна. Клиническое обследование членов семьи больного, а также электрофизиологическое и биохимическое исследования позволяют выявить малые признаки болезни. Плече-лопаточно-лицевая миодистрофия Ландузи — Дежерина. Тип наследования аутосомно-доминантный, имеются спорадические случаи. Заболевание начинается в возрасте 12—20 лет. Характерны слабость и атрофия мышц лица, плечевого пояса, проксимальных отделов рук, следствием чего являются амимия, отставание лопаток (крыловидные лопатки) при попытке поднять руки вверх, деформация грудной клетки и позвоночника. Генерализация патологического процесса продолжается 10—15 лет, он постепенно распространяется на мышцы тазового пояса, проксимальных и дистальных отделов ног. В некоторых случаях развиваются также атрофии мышц бедер и голеней. Псевдогипертрофия икроножных, дельтовидных, лицевых мышц выражена умеренно. Рефлексы могут быть долгое время сохранены. Течение медленно прогрессирующее, больные длительное время сохраняют трудоспособность. Интенсивные физические нагрузки ведут к быстрому прогрессированию заболевания.Дистальная миодистрофия Говерса относится к редким заболеваниям. Тип наследования аутосомно-доминантный, имеются спорадические случаи. Проявляется в возрасте 30—60 лет. Характерными симптомами являются слабость и атрофии мышц голени и стоп, снижение ахилловых и коленных рефлексов. Генерализация процесса с развитием атрофий кистей и проксимальных отделов конечностей происходит в течение 5—10 лет. У больных, как правило, выражена кардиомиопатия, приводящая к летальному исходу.
Офтальмоплегическая и окулофарингеальная (глазо-глоточная) формы. Тип наследования аутосомно-доминантный, имеются спорадические случаи. Возраст начала заболевания может быть разным. Характерны медленно нарастающая слабость и атрофия глазодвигательных мышц, ограничение движений глазных яблок, опущение верхнего века. Поражение глазных мышц может быть изолированным или сочетаться с атрофией мышц лица, глотки (что приводит к затруднению глотания), поражением мышц конечностей. Течение медленно прогрессирующее. Диагноз. Д. м. п. может быть поставлен в амбулаторных условиях. Обследование в стационаре необходимо в случаях редких форм или для проведения дифференциального диагноза. Во всех случаях обнаружения у больного мышечной слабости и атрофии, двигательных нарушений прогрессирующего характера необходимо изучить семейный анамнез, обследовать родственников (гетерозиготных носителей) для выявления у них малых признаков заболевания. Дифференциальный диагноз следует проводить с врожденными миопатиями (Миопатии), характерными признаками которых являются диффузная мышечная гипотония, гипотрофия. Д. м. п. следует также дифференцировать с вторичными миопатическими синдромами, которые возникают на фоне воспалительных, сосудистых, токсических, метаболических процессов, или обусловлены воздействием физических факторов. Лечение патогенетическое и симптоматическое. Комплексный курс лекарственной терапии включает назначение аминокислот (глутаминовой кислоты, метионина), пирацетама, витаминов (А, В, С, D, Е, кофермента О), сосудорасширяющих препаратов (ксантинали никошнат), никотиновой кислоты, средств, корригирующих энергетические процессы в мышцах (АТФ, АДФ), антихолинэстеразных препаратов (прозерина, сангвиритрина, пиридостигмина бромида), препаратов калия и кальция (калия хлорида, кальция глюконата), анаболических гормонов (неробола, ретаболила). Последние назначают лицам мужского пола в случаях мышечной кахексии для увеличения объема и массы мышц. ЛФК, массаж применяют строго дозированно по индивидуальным схемам. Благоприятный эффект оказывает электростимуляция мышц. Ортопедические мероприятия направлены на профилактику ретракции и контрактур. Рекомендуется преимущественно молочно-растительная диета (фрукты, овощи, продукты козьего молока, орехи, мед). Во всех случаях заболевания комплексное лечение способствует стабилизации процесса и улучшению самочувствия больных. Библиогр.: Бадалян Л.О. Детская неврология, с. 400, М., 1984; Бадалян Л.О. и Скворцов И.А. Клиническая электронейромиография, с. 166, М., 1986; Гусманова-Петрусевич И. Мышечные заболевания, пер. с польск., Варшава, 1971; Гехт Б.М. и Ильина Н.А. Нервно-мышечные болезни, с. 57, М., 1982; Клинико-электронейромиографическое изучение нервно-мышечных заболеваний и синдромов, под ред. Л.О. Бадаляна и И.А. Скворцова, с. 3, М., 1982; Лечение нервных болезней, под ред. В.К. Видерхольда, пер. с англ., с. 73, М., 1984; Миопатии, под рея. С. Божинова и Г. Гылабова, пер. с болг. София, 1977.Мышечная дистрофия — Диагностика и лечение
Диагноз
Ваш врач, скорее всего, начнет с истории болезни и медицинского осмотра.
После этого ваш врач может порекомендовать:
- Ферментные тесты. Поврежденные мышцы выделяют в кровь ферменты, такие как креатинкиназа (КК). У человека, не получившего травм, высокий уровень в крови CK указывает на мышечное заболевание.
- Генетическое тестирование. Образцы крови могут быть исследованы на наличие мутаций в некоторых генах, вызывающих типы мышечной дистрофии.
- Биопсия мышц. Небольшой кусок мышцы можно удалить через разрез или с помощью полой иглы. Анализ образца ткани позволяет отличить мышечные дистрофии от других мышечных заболеваний.
- Кардиомониторинг-пробы (электрокардиография и эхокардиограмма). Эти тесты используются для проверки функции сердца, особенно у людей с диагнозом миотоническая мышечная дистрофия.
- Тесты для мониторинга легких. Эти тесты используются для проверки функции легких.
- Электромиография. В исследуемую мышцу вводится игла электрода. Электрическая активность измеряется, когда вы расслабляетесь и когда вы мягко напрягаете мышцы. Изменения в характере электрической активности могут подтвердить мышечное заболевание.
Лечение
Хотя нет лекарства от любой формы мышечной дистрофии, лечение некоторых форм болезни может помочь продлить время, в течение которого больной человек может оставаться подвижным, и укрепить сердечную и легочную мышцу.Испытания новых методов лечения продолжаются.
Люди с мышечной дистрофией должны находиться под наблюдением в течение всей жизни. Их команда по уходу должна включать невролога, специализирующегося в области нервно-мышечных заболеваний, специалиста по физиотерапии и реабилитации, а также физиотерапевтов и терапевтов.
Некоторым людям также может потребоваться специалист по легким (пульмонолог), кардиолог (кардиолог, специалист по сну, специалист по эндокринной системе (эндокринолог), хирург-ортопед и другие специалисты.
Варианты лечения включают лекарства, физиотерапию и трудотерапию, а также хирургические и другие процедуры. Постоянные оценки ходьбы, глотания, дыхания и функции рук позволяют терапевтической бригаде корректировать лечение по мере прогрессирования заболевания.
Лекарства
Ваш врач может порекомендовать:
- Кортикостероиды, такие как преднизон и дефлазакорт (Эмфлаза), которые могут повысить мышечную силу и замедлить прогрессирование определенных типов мышечной дистрофии.Но длительное употребление этих препаратов может вызвать увеличение веса и ослабление костей, увеличивая риск переломов.
Новые лекарства включают этеплирсен (Exondys 51), первое лекарство, одобренное Управлением по контролю за продуктами и лекарствами (FDA) специально для лечения некоторых людей с мышечной дистрофией Дюшенна. Условно утвержден в 2016 году.
В 2019 году FDA одобрило голодирсен (Vyondys 53) для лечения некоторых людей с дистрофией Дюшенна, у которых есть определенная генетическая мутация.
- Сердечные препараты, такие как ингибиторы ангиотензин-превращающего фермента (АПФ) или бета-блокаторы, если мышечная дистрофия повреждает сердце.
Терапия
Некоторые виды терапии и вспомогательных устройств могут улучшить качество, а иногда и продолжительность жизни людей с мышечной дистрофией. Примеры включают:
- Диапазон движений и упражнения на растяжку. Мышечная дистрофия может ограничивать гибкость и подвижность суставов.Конечности часто втягиваются внутрь и фиксируются в этом положении. Упражнения на диапазон движений могут помочь сохранить суставы максимально гибкими.
- Упражнение. Аэробные упражнения с малой нагрузкой, такие как ходьба и плавание, могут помочь сохранить силу, подвижность и общее состояние здоровья. Также могут быть полезны некоторые виды укрепляющих упражнений. Но сначала важно поговорить с врачом, потому что некоторые виды упражнений могут быть вредными.
- Подтяжки. Подтяжки помогают поддерживать растяжение и гибкость мышц и сухожилий, замедляя прогрессирование контрактур.Подтяжки также могут способствовать подвижности и функционированию, поддерживая ослабленные мышцы.
- Средства передвижения. Трости, ходунки и инвалидные коляски помогают сохранять подвижность и независимость.
- Помощь при дыхании. По мере ослабления дыхательных мышц устройство для апноэ во сне может помочь улучшить доставку кислорода в ночное время. Некоторым людям с тяжелой мышечной дистрофией необходимо использовать аппарат, который нагнетает воздух в легкие и выходит из них (вентилятор).
Хирургия
Может потребоваться операция для исправления контрактур или искривления позвоночника, которые в конечном итоге могут затруднить дыхание.Функцию сердца можно улучшить с помощью кардиостимулятора или другого сердечного устройства.
Профилактика респираторных инфекций
Респираторные инфекции могут стать проблемой при мышечной дистрофии. Поэтому важно сделать прививку от пневмонии и быть в курсе прививок от гриппа. Старайтесь избегать контакта с детьми или взрослыми, у которых есть явная инфекция.
Клинические испытания
Изучите исследования клиники Мэйо, в которых тестируются новые методы лечения, вмешательства и тесты как средства предотвращения, обнаружения, лечения или управления этим заболеванием.
Помощь и поддержка
Диагностика мышечной дистрофии может быть чрезвычайно сложной задачей. Чтобы помочь вам справиться, найдите с кем поговорить. Вы можете чувствовать себя комфортно, обсуждая свои чувства с другом или членом семьи, или вы можете предпочесть встречу с официальной группой поддержки.
Если у вашего ребенка мышечная дистрофия, спросите своего врача, как обсудить это прогрессирующее состояние с вашим ребенком.
Подготовка к приему
Возможно, вас направят к врачу, который специализируется на диагностике и лечении мышечной дистрофии.
Что вы можете сделать
- Запишите признаки и симптомы у вас или вашего ребенка и когда они начались.
- Принесите фотографии или видеозаписи, чтобы показать врачу симптомы, которые вас беспокоят.
- Запишите основную медицинскую информацию, включая другие состояния.
- Составьте список всех лекарств, витаминов и добавок, которые вы или ваш ребенок принимаете, включая дозы.
- Сообщите врачу, был ли у кого-нибудь в вашей семье диагноз мышечной дистрофии.
Вопросы, которые следует задать врачу или врачу вашего ребенка
- Какая наиболее вероятная причина этих признаков и симптомов?
- Какие тесты нужны?
- Каковы возможные осложнения этого состояния?
- Какие процедуры вы рекомендуете?
- Каковы долгосрочные перспективы?
- Вы рекомендуете, чтобы наша семья встретилась с генетическим консультантом?
Не стесняйтесь задавать другие вопросы во время приема.
Чего ожидать от врача
Ваш врач, скорее всего, задаст вам вопросы, например:
- Симптомы ухудшаются?
- Что их успокаивает?
- Что делает их еще хуже?
- Планируете ли вы завести еще детей?
31 января 2020 г.
Какие условия могут вызвать мышечное истощение?
Мышечное истощение — это потеря мышечной массы из-за ослабления и сокращения мышц.Существует несколько возможных причин истощения мышц, в том числе определенные заболевания, такие как боковой амиотрофический склероз.
Симптомы мышечной атрофии зависят от тяжести потери мышечной массы, но типичные признаки и симптомы включают:
- снижение мышечной силы
- нарушение способности выполнять физические нагрузки
- уменьшение размера мышц
Диагностика обычно возникает после изучения истории болезни и физического осмотра.Причина истощения мышц иногда очевидна. В других случаях врачу может потребоваться провести дополнительные тесты для подтверждения диагноза.
Медицинские состояния, которые могут вызвать мышечное истощение, включают следующие:
Боковой амиотрофический склероз (БАС) — прогрессирующее заболевание, поражающее нервные клетки по всему телу.
Обычно нервные клетки в головном и спинном мозге посылают мышцам сигнал о движении.
У людей с БАС нервные клетки, контролирующие произвольные движения, умирают и перестают посылать сигналы, позволяющие двигаться.В конце концов, из-за отсутствия использования мышцы атрофируются.
Врачи не знают, что вызывает БАС.
Узнайте больше о ALS здесь.
Рассеянный склероз (РС) — это тип аутоиммунного заболевания, при котором поражается миелин, окружающий нервные волокна.
Состояние вызывает повреждение нервов, которое, в свою очередь, влияет на мышцы. Поврежденные нервы теряют способность запускать мышечные движения, что приводит к атрофии.
Степень повреждения влияет на скорость потери мышечной массы.
Узнайте больше о рассеянном склерозе здесь.
Спинальная мышечная атрофия — это состояние, похожее на мышечную дистрофию.
Заболевание является генетическим и возникает из-за потери двигательных нейронов — клеток, контролирующих мышцы. Мышцы тела постепенно ослабевают.
Хотя спинальная мышечная атрофия ослабляет большинство мышц тела, обычно наиболее сильно поражаются мышцы, расположенные ближе к центру тела.
Узнайте больше о спинальной мышечной атрофии здесь.
Некоторые другие причины мышечной атрофии, которые сами по себе не являются медицинскими состояниями, включают:
Длительное бездействие
Продолжительное бездействие, например постельный режим, может привести к потере мышечной массы. Постельный режим может быть необходим из-за травм или болезней, из-за которых человек не может двигаться.
Согласно исследованиям, мышечное истощение может развиться в течение 10 дней у здоровых пожилых людей, находящихся в постельном режиме. Из-за истощения мышц в течение первой недели может произойти снижение мышечной силы на 40%.
Недоедание
Люди с недоеданием получают значительно неадекватное питание, и это может вызвать потерю мышечной массы, что приводит к истощению мышц.
Недоедание имеет множество возможных причин, включая нервную анорексию, рак и постоянную тошноту.
Старение
Потеря мышечной массы происходит постепенно из-за старения.
Авторы исследования 2013 года отметили, что значительные изменения в мышечной массе ног происходят в возрасте после 50 лет, когда типичная потеря мышечной массы составляет 1-2% в год.
Они также отметили исследования, показывающие, что в возрасте от 20 до 80 лет средний человек теряет 35-40% мышечной массы ног.
Лечение мышечной атрофии жизненно важно для общего благополучия человека.
Мышечное истощение возникает при многих заболеваниях. Согласно исследованию 2017 года, истощение мышц способствует ухудшению прогноза при таких заболеваниях, как сердечная недостаточность, сепсис и рак.
Лечение может частично зависеть от основного состояния, приводящего к потере мышечной массы.В некоторых случаях лечение болезни может предотвратить дальнейшее истощение мышц и помочь изменить состояние.
Дополнительные варианты лечения могут включать:
Упражнения
Упражнения для наращивания силы — один из основных способов предотвращения и лечения мышечной атрофии. Вид деятельности, которую рекомендуют врачи, будет зависеть от причины атрофии. Например, определенные основные условия могут ограничивать выполнение определенных упражнений.
Сфокусированная ультразвуковая терапия
Сфокусированная ультразвуковая терапия — относительно новое лечение мышечной атрофии.Он включает в себя направление лучей высокочастотных звуковых волн на определенные участки тела. Звуковые волны стимулируют сокращение мышц, что может помочь уменьшить их потерю.
Диетотерапия
Правильное питание помогает телу наращивать и сохранять мышцы. Принятие диеты, обеспечивающей достаточное количество калорий, белка и других питательных веществ, способствующих развитию мышц, может помочь в лечении мышечной атрофии.
Физиотерапия
Физическая терапия может включать в себя различные методы предотвращения мышечного истощения.Терапевты могут порекомендовать определенные упражнения в зависимости от состояния человека.
Физиотерапия также полезна, если человек находится в постельном режиме. Терапевты могут выполнять пассивные упражнения, если человек не может двигаться. В этом типе упражнений терапевт двигает руками и ногами для тренировки мышц.
Мышечная атрофия включает потерю или атрофию мышц и обычно происходит постепенно. Это может произойти из-за множества состояний, включая БАС, мышечную дистрофию и РС.
Поскольку истощение мышц может повлиять на силу человека и его способность выполнять повседневную деятельность, оно может значительно снизить качество его жизни.
Как можно скорее вылечить это заболевание, чтобы предотвратить или замедлить значительную потерю мышечной массы. Всем, кто думает, что у них может быть мышечная атрофия, следует обратиться к врачу.
В некоторых случаях можно обратить вспять мышечное истощение, но это может занять время. Когда мышечное истощение необратимо, лечение может, по крайней мере, замедлить потерю мышц.Лечение может включать сочетание упражнений, изменений в питании и физиотерапии.
Мышечная атрофия: причины, симптомы и лечение
Термин «мышечная атрофия» относится к потере мышечной ткани. Атрофированные мышцы кажутся меньше нормальных. Отсутствие физической активности из-за травмы или болезни, плохого питания, генетики и определенных заболеваний может способствовать атрофии мышц.
Атрофия мышц может возникнуть после длительного бездействия. Если мышца не используется, тело в конечном итоге разрушает ее, чтобы сохранить энергию.
Атрофия мышц, развивающаяся из-за бездействия, может возникнуть, если человек остается неподвижным, пока он выздоравливает после болезни или травмы. Регулярные упражнения и физиотерапия могут обратить вспять эту форму мышечной атрофии.
Люди могут лечить мышечную атрофию, изменив образ жизни, попробовав физиотерапию или перенеся операцию.
В этой статье мы рассмотрим некоторые другие причины, симптомы и методы лечения атрофии мышц.
Многие факторы могут вызвать атрофию мышц, в том числе:
Плохое питание
Плохое питание может привести к многочисленным заболеваниям, включая атрофию мышц.
В частности, Международный фонд остеопороза предупреждает, что диета с низким содержанием постного белка, фруктов и овощей может привести к снижению мышечной массы.
Атрофия мышц, связанная с неправильным питанием, может развиться в результате заболеваний, которые ухудшают способность организма усваивать питательные вещества, например:
Кахексия — сложное метаболическое состояние, которое вызывает сильную потерю веса и атрофию мышц. Кахексия может развиваться как симптом другого основного заболевания, такого как рак, ВИЧ или рассеянный склероз (РС).
У людей с кахексией может наблюдаться значительная потеря аппетита или непреднамеренная потеря веса, несмотря на потребление большого количества калорий.
Возраст
По мере того, как человек становится старше, его организм вырабатывает меньше белков, которые способствуют росту мышц. Это сокращение доступного белка вызывает сокращение мышечных клеток, что приводит к состоянию, называемому саркопенией.
Согласно отчету Управления по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA), саркопения поражает до трети людей в возрасте 60 лет и старше.
Помимо снижения мышечной массы, саркопения может вызывать следующие симптомы:
- слабость или хрупкость
- плохое равновесие
- трудности с движением
- снижение выносливости
Потеря мышечной массы может быть неизбежным результатом естественного процесс старения. Однако это может увеличить риск травм и негативно повлиять на общее качество жизни человека.
Генетика
Спинальная мышечная атрофия (СМА) — это генетическое заболевание, которое вызывает потерю двигательных нервных клеток и атрофию мышц.
Существует несколько различных типов SMA, которые попадают в следующие категории:
- SMA, связанная с хромосомой 5 : Эти типы SMA возникают из-за мутации в генах SMN1 на хромосоме 5. Мутации приводят к дефицит белка выживания мотонейрона. СМА обычно развивается в детстве, но может развиться в любой момент жизни.
- SMA, не связанная с хромосомой 5
Мышечная дистрофия относится к группе прогрессирующих состояний, которые вызывают потерю мышечной массы и слабость.
Мышечная дистрофия возникает, когда один из генов, участвующих в производстве белка, мутирует. Человек может унаследовать генетические мутации, но многие из них возникают естественным образом по мере развития эмбриона.
Состояние здоровья
Поделиться на PinterestАтрофированные мышцы меньше здоровых.Изображение предоставлено: OpenStax, 2016.
Заболевания и хронические состояния, которые могут способствовать атрофии мышц, включают:
- Боковой амиотрофический склероз (БАС) : БАС, также называемый болезнью Лу Герига, включает несколько типов, которые повреждают двигательные нервные клетки, которые повреждают двигательные нервные клетки. контролировать мышцы.
- MS : Это хроническое заболевание возникает, когда иммунная система организма атакует центральную нервную систему, вызывая опасное воспаление нервных волокон.
- Артрит : Артрит относится к воспалению суставов, которое вызывает боль и скованность. Артрит может серьезно ограничить подвижность человека, что может привести к неиспользованию мышц и атрофии.
- Миозит : Термин миозит относится к воспалению мышц. Это состояние вызывает мышечную слабость и боль.У людей может развиться миозит после вирусной инфекции или как побочный эффект аутоиммунного заболевания.
- Полиомиелит : это инфекционное заболевание поражает нервную систему. Это вызывает симптомы гриппа и может привести к необратимому параличу.
Неврологические проблемы
Травма или состояние могут повредить нервы, контролирующие мышцы, что приведет к состоянию, которое называется нейрогенной мышечной атрофией.
Когда это развивается, мышцы перестают сокращаться, потому что они больше не получают сигналы от нерва.
Симптомы мышечной атрофии сильно различаются в зависимости от причины и тяжести мышечной потери.
В дополнение к уменьшению мышечной массы, симптомы атрофии мышц включают:
- , когда одна рука или нога заметно меньше других
- испытывает слабость в одной конечности или обычно
- испытывает трудности с равновесием
- остается неактивным в течение длительный период
Лечение атрофии мышц различается в зависимости от степени потери мышц и наличия каких-либо сопутствующих заболеваний.
Лечение основного состояния, вызывающего атрофию мышц, может помочь замедлить прогрессирование потери мышц.
К лечению атрофии мышц относятся:
Физическая терапия
Поделиться на Pinterest Физическая терапия может помочь улучшить подвижность людей с атрофией мышц.Физическая терапия включает выполнение определенных упражнений на растяжку и упражнения с целью предотвращения неподвижности. Физическая терапия предлагает следующие преимущества людям с атрофией мышц:
- предотвращение неподвижности
- увеличение мышечной силы
- улучшение кровообращения
- уменьшение спастичности, вызывающей непрерывное сокращение мышц
Функциональная электрическая стимуляция
Функциональная электрическая стимуляция (FES ) — еще одно эффективное лечение атрофии мышц.Он включает использование электрических импульсов для стимуляции сокращения пораженных мышц.
Во время FES обученный техник прикрепляет электроды к атрофированной конечности. Электроды передают электрический ток, который вызывает движение в конечности.
Сфокусированная ультразвуковая терапия
Этот метод доставляет лучи ультразвуковой энергии к определенным участкам тела. Лучи стимулируют сокращение атрофированной мышечной ткани.
Эта новая технология находится в стадии разработки и еще не вошла в стадию клинических испытаний.
Хирургия
Хирургические процедуры могут улучшить функцию мышц у людей, атрофия мышц которых связана с неврологическими состояниями, травмами или недоеданием.
Атрофия мышц, или истощение мышц, характеризуется значительным укорочением мышечных волокон и потерей общей мышечной массы.
Несколько факторов могут способствовать атрофии мышц, например:
- длительное пребывание в неподвижности из-за болезни или травмы
- возраст
- недоедание
- генетика
- неврологические проблемы
- определенные заболевания, такие как артрит, миозит , ALS и MS
Варианты лечения будут зависеть от каждого отдельного случая, но они могут включать физиотерапию, диетическое вмешательство или хирургическое вмешательство.
Причины, симптомы и варианты лечения
Медицинский осмотр на сайте Drugs.com. Последнее обновление: 30 октября 2020 г.
Что такое мышечная дистрофия?
Мышечная дистрофия (МД) — это группа заболеваний, при которых мышцы тела становятся все более слабыми. Мышечная дистрофия — это наследственное заболевание.
Типы мышечной дистрофии включают:
- Дистрофия Дюшенна — это самый тяжелый тип мышечной дистрофии.Тоже самое распространенное. При дистрофии Дюшенна мышцы производят аномально низкий уровень дистрофина. Дистрофин — это мышечный белок. Когда уровень дистрофина низкий, мембраны вокруг мышечных клеток становятся слабыми. Они легко рвутся. В конце концов мышечные волокна отмирают. Дистрофия Дюшенна в первую очередь поражает мальчиков. У женщин симптомы дистрофии Дюшенна обычно отсутствуют. Но они могут передать это своим детям.
- Мышечная дистрофия Беккера — Подобно дистрофии Дюшенна, тип Беккера влияет на выработку дистрофина и встречается у мужчин.Однако болезнь встречается гораздо реже, чем Дюшенна. И это вызывает более легкие симптомы. У некоторых людей есть форма болезни, которая находится между типами Дюшенна и Беккера.
- Миотоническая дистрофия — Миотоническая дистрофия может возникать у мужчин и женщин. Это наиболее распространенная форма MD у взрослых. Симптомы часто начинаются в подростковом возрасте.
- Конечностно-поясная мышечная дистрофия — Конечностно-поясная мышечная дистрофия. MD поражает мышцы плеч и бедер. Он включает в себя несколько разных болезней.Он может передаваться по наследству как мужчинам, так и женщинам.
- Facioscapulohumeral мышечная дистрофия — Эта форма MD встречается у мужчин и женщин. Типичная слабость лицевых мышц и плеч. Facioscapulohumeral MD часто поражает нескольких членов одной семьи. Однако симптомы различаются по интенсивности. У членов семьи могут быть такие легкие симптомы, что они не подозревают о проблемах с мышцами.
Есть как минимум две другие формы болезни.Это врожденная мышечная дистрофия и дистрофия Эмери-Дрейфуса. Emery-Dreifuss MD вызывает слабость плеча и голени, а также нарушение сердечной деятельности. Некоторые формы врожденной мышечной дистрофии связаны со снижением психической функции.
Симптомы
Симптомы MD различаются в зависимости от конкретной формы болезни. Возраст начала также варьируется.
- Дистрофия Дюшенна — Симптомы обычно появляются в возрасте от 2 до 4 лет.Мышцы ног становятся все слабее. В результате ребенок часто падает и с трудом поднимается с земли. Ребенку также трудно нормально ходить или бегать. Икры постепенно увеличиваются в размерах, даже когда ноги становятся слабее. Также наблюдается некоторая слабость мышц рук и шеи. Но обычно это не так серьезно, как слабость в ногах. К 12 годам многие пациенты не могут самостоятельно ходить и нуждаются в инвалидной коляске. Кроме того, у некоторых людей с дистрофией Дюшенна есть умственные нарушения.У них также могут быть проблемы с сердцем, связанные с MD.
- Мышечная дистрофия Беккера — Симптомы аналогичны симптомам дистрофии Дюшенна, но они более легкие. Они также начинаются позже, обычно в возрасте от 5 до 15 лет.
- Миотоническая дистрофия — Основной симптом — ненормально продолжительные мышечные сокращения и трудности с расслаблением мышц. Это называется миотонией мышц. В основном поражаются руки, запястья и язык. Он может развиться вскоре после рождения или уже в раннем взрослом возрасте.Также наблюдается истощение и ослабление лицевых мышц, мышц шеи и мышц запястий, пальцев и лодыжек. Если задействованы мышцы языка и горла, у пациента могут возникнуть проблемы с речью и затруднение глотания. Если задействованы диафрагма и грудная мышца, могут возникнуть проблемы с дыханием.
- Мышечная дистрофия конечностей и пояса — Симптомы начинаются в позднем детстве или в раннем взрослом возрасте. Они включают прогрессирующую мышечную слабость в плечах и бедрах.Проблемы с дыханием возникают, если задействована диафрагма. Если болезнь поражает сердечную мышцу, возможна сердечная недостаточность или нарушение сердечного ритма.
- Facioscapulohumeral мышечная дистрофия — Симптомы могут возникать в младенчестве, позднем детстве или в раннем взрослом возрасте. Обычно первым признаком является слабость лица. Трудно улыбаться, свистеть и закрывать глаза. Позже возникают трудности с поднятием рук или сгибанием запястий и / или лодыжек.
Диагностика
Ваш врач изучит вашу (или вашего ребенка) историю болезни и симптомы.Врач спросит об истории развития. Это включает возраст первого сидения, стояния и ходьбы. Вас также спросят об истории инфекций и любых серьезных травмах.
Ваш врач осмотрит вас или вашего ребенка. Он или она будет искать мышечную слабость, истощение и миотонию.
Врач проведет неврологическое обследование на предмет нервных расстройств.
Для подтверждения диагноза мышечной дистрофии врач может заказать:
- Анализы крови. Измеряют уровни определенного мышечного фермента.
- Биопсия мышцы. Крошечный образец мышцы будет взят для исследования в лаборатории.
- Электромиограмма (ЭМГ). Этот тест регистрирует электрическую активность мышц.
- Генетический анализ крови . Это проверяет наличие определенных наследственных аномалий в генетическом материале.
Ожидаемая продолжительность
Все формы мышечной дистрофии со временем ухудшаются.Они сохраняются на протяжении всей жизни.
Профилактика
Нет способа предотвратить мышечную дистрофию.
Генетическое тестирование может помочь определить шансы иметь ребенка с MD. Обычно это простой анализ крови.
Лечение
Нет лекарства от мышечной дистрофии и нет способа остановить ее ухудшение. Пациентам предоставляется поддерживающая терапия, ортезы для ног и физиотерапия, чтобы помочь им функционировать в повседневной жизни.
Особенно важно, чтобы конечности были растянуты. Это помогает предотвратить сжатие сухожилий и мышц. Когда возникает стеснение сухожилий (контрактуры), можно делать операцию.
При поражении грудных мышц в MD, респираторная терапия может использоваться для отсрочки дыхания.
Людям с MD также назначают диетическую терапию, соответствующую возрасту. Это помогает предотвратить ожирение. Ожирение особенно вредно для пациентов с мышечной дистрофией.Это создает дополнительную нагрузку на их слабые мышцы. К сожалению, MD увеличивает риск ожирения, потому что физические ограничения не позволяют людям с MD заниматься спортом.
Некоторые формы MD затрагивают сердце. Пациентам с этими формами следует оценить функцию сердца и ритм. Могут быть полезны лекарства, улучшающие работу сердца и ритм. Кардиостимулятор помогает поддерживать нормальную частоту сердечных сокращений.
У мальчиков с дистрофией Дюшенна кортикостероиды могут временно предотвратить обострение болезни.Однако некоторые люди не переносят это лекарство.
Мощные лекарства, подавляющие иммунную систему, могут помочь некоторым пациентам. Но их использование является спорным и несколько рискованно.
У пациентов с миотонической дистрофией мышечные сокращения можно лечить с помощью лекарств.
Когда звонить профессионалу
Позвоните своему врачу, если вы заметили мышечную слабость в любом месте своего тела. Также звоните, если у вас проблемы с четкой речью или нормальным глотанием.
Если вы родитель, позвоните врачу вашего ребенка, если у вашего ребенка:
- Необычно частые падения
- Проблемы с подъемом с земли
- Проблемы при ходьбе
- Неисправность при работе
Прогноз
Прогноз варьируется в зависимости от:
- Тип мышечной дистрофии
- Как быстро это прогрессирует
У некоторых пациентов наблюдаются только легкие симптомы при нормальной продолжительности жизни.Другие имеют тяжелые симптомы и умирают в молодом возрасте.
Узнать больше о мышечной дистрофии
Сопутствующие препараты
IBM Watson Micromedex
Справочник клиники Mayo
Клинические коды МКБ-10 CM (внешние)
Руководства Medicine.com (внешние)
Внешние ресурсы
Национальный институт неврологических расстройств и инсульта
http://www.ninds.nih.gov/
Ассоциация мышечной дистрофии
http: // www.mdausa.org/
Родительский проект по исследованию мышечной дистрофии
http://www.parentdmd.org/
Дополнительная информация
Всегда консультируйтесь со своим врачом, чтобы убедиться, что информация, отображаемая на этой странице, применима к вашим личным обстоятельствам.
Заявление об отказе от ответственности за медицинское обслуживание
Что такое мышечная дистрофия, симптомы и лечение мышечной дистрофии
Что такое мышечная дистрофия?
Многие ищут ответ на вопрос, что такое мышечная дистрофия? Мышечная дистрофия — это мышечное заболевание, которое возникает из-за дефектных генов.Мышцы помогают нам двигать телом. Есть некоторые отсутствующие гены или неверная информация в генах, которые препятствуют образованию белков, необходимых для здорового функционирования мышц. Это генетическое заболевание, поэтому оно присутствует во время родов. Это неинфекционное заболевание. При этом заболевании мышцы становятся слабыми, из-за чего человек не может выполнять повседневные действия, в которых задействованы мышцы. Людям, страдающим мышечной дистрофией, трудно ходить, стоять или сидеть.Существуют разные типы мышечных дистрофий, которые могут поражать людей разных возрастных групп в разные периоды времени. При этом заболевании мышцы различных частей тела со временем слабеют и могут вызывать проблемы при выполнении повседневных дел.
Причины мышечной дистрофии
Все случаи мышечной дистрофии передаются по наследству от родителей из-за дефектных генов. Есть определенный ген, вызывающий мышечную дистрофию. Симптомы мышечной дистрофии зависят от гена и хромосомы, на которой расположен конкретный ген.Дефектный ген, присутствующий в хромосоме, не производит необходимый белок или мышцы, которым требуется конкретный белок. Поэтому родители должны сдать анализ на болезнь, прежде чем пытаться зачать ребенка. Это поможет снизить вероятность рождения детей с мышечной дистрофией.
Купить Пакет для мышечной дистрофии (Musc. Dy)
Симптомы мышечной дистрофии
Симптомы мышечной дистрофии зависят от возраста начала и типа. Некоторые из общих симптомов мышечной дистрофии:
- Люди с мышечной дистрофией не могут устойчиво ходить, у них могут быть частые падения
- Слабость в мышцах, из-за которой человеку трудно заниматься обычной деятельностью
- Скованность в суставах может возникать из-за слабости мышц и суставов
- Пациент может легко устать из-за слабой мускулатуры
Лечение мышечной дистрофии
Пакет для детей с мышечной дистрофией и детей с ограниченными возможностями — это отличный набор травяных и природных средств, которые могут помочь предотвратить признаки и симптомы мышечной дистрофии.Этот пакет лечебных средств является естественным средством лечения мышечной дистрофии. Все средства, содержащиеся в этой упаковке, безопасны и натуральны и не вызывают побочных эффектов при регулярном длительном приеме для избавления от мышечной слабости. Эти лечебные травы обеспечивают питание мышц и предотвращают боли и слабость в мышцах. Этот пакет для лечения мышечной дистрофии и детей с ограниченными возможностями состоит из следующих лечебных трав:
Дивья Экангвеер Рас: Это лечебное средство на травах показано при мышечных болях.Это отличное средство, которое питает мышцы и помогает в лечении боли и жесткости суставов. Он предотвращает признаки и симптомы мышечной дистрофии, снабжая мышцы питанием.
Divya Godanti Bhasm: Это также отличное средство от мышечной дистрофии. Он снимает боль без побочных эффектов. Это лечебное средство на травах, которое увеличивает силу мышц и суставов и помогает выполнять упражнения.
Дивья Гилой Sat: Это лечебное средство на травах лучше всего подходит для людей, страдающих любым видом мышечных заболеваний. Он обеспечивает питание мышц, а также помогает в образовании белка, необходимого для нормального функционирования мышц.
Divya Swarnamakshik Bhasm: Это отличный травяной продукт, обеспечивающий безопасное лечение мышечных заболеваний. Это увеличивает силу мышц и помогает в движении мышц. Это помогает в нормальном развитии детей, страдающих мышечными нарушениями.
Дивья Расрадж Рас: Это лечебное средство на травах подходит для людей, страдающих от болей в суставах и мышечной слабости. Это помогает легко использовать мышцы, не чувствуя боли. Он увеличивает приток кислорода и крови к мышечным клеткам и снимает скованность и боль в мышцах.
Дивья Васант Кусумакар Рас: Обеспечивает правильное питание мышечных клеток и помогает избавиться от мышечных болей. Это помогает в нормальном развитии, и дети могут легко использовать свои мышцы во время бега, игры, письма или ходьбы.Это лечебное средство на травах увеличивает силу и энергию мышц, обеспечивая быстрое облегчение.
Дивья Мукта Пишти: Это натуральное лечебное средство на травах, которое полезно для лечения мышечных заболеваний, таких как мышечная дистрофия. Его можно начинать в раннем возрасте, чтобы предотвратить долгосрочные осложнения мышечной дистрофии. Это натуральное средство, обеспечивающее питание мышечных клеток для их эффективного функционирования.
Дивья Шиладжит Расаян Вати: Это лечебное средство на травах помогает увеличить силу мускулов.Он увеличивает энергию клеток электронного тела и снимает мышечные боли. Это помогает облегчить работу мышц, а также стимулирует организм вырабатывать необходимые белки для развития мышц.
Дивья Трайодашанг Гуггулу: Это лечебное средство на травах полезно при усталости и слабости, которые присутствуют у людей, страдающих мышечными дистрофиями. Это помогает повысить энергию и силу мышц, чтобы можно было легко выполнять повседневные дела.
Дивья Чандрапрабха Вати: Это лечебное средство на травах подходит для людей, страдающих мышечными дистрофиями. Это естественное средство, которое обеспечивает клетки питательными веществами для правильного функционирования. Он увеличивает силу и уменьшает боль и слабость в мышцах. Он обеспечивает полное избавление от мышечных болей за счет повышения энергии.
Divya Ashwagandha Churna: Это прекрасное лечебное средство на травах, которое действует на мышцы пищеварительной системы и помогает в правильном переваривании пищи.Он помогает в правильном усвоении, а также увеличивает мышечную силу, снимая боли и жесткость мышц.
.