Врожденная ихтиозиформная эритродермия Брока. Что такое Врожденная ихтиозиформная эритродермия Брока?
ВАЖНО
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Врожденная ихтиозиформная эритродермия Брока – одна из форм врождённой ихтиозиформной эритродермии, отличительной особенностью которой является преобладание явлений гиперкератоза над остальными клиническими симптомами, отсутствие высыпаний на слизистых оболочках. Первичными элементами являются эритематозные пятна, обладающие тенденцией к периферическому росту, с гиперкератотическими наслоениями или небольшими везикулами. Решающим моментом в диагностике является семейный анамнез и клиника, в качестве подтверждения проводится гистологическое исследование. Лечение симптоматическое – увлажнение и смягчение кожи, периодические курсы витамина А, кортикостероидов.
Лечение симптоматическое – увлажнение и смягчение кожи, периодические курсы витамина А, кортикостероидов.
- Причины
- Симптомы
- Диагностика
- Лечение врожденной ихтиозиформной эритродермии Брока
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Врожденная ихтиозиформная эритродермия Брока – редкая разновидность возникающего с момента рождения универсального гиперкератоза. Впервые заболевание описал французский дерматолог Луи Брок в 1902 году. Большой практический опыт и значительное количество клинических наблюдений дали ученому возможность первым описать ряд дерматозов, известных и сегодня под его именем: псевдопелада Брока, люпоидный сикоз Брока, врожденная ихтиозиформная эритродермия Брока. За характерный внешний вид кожи при врожденной ихтиозиформной эритродермии исследователь метко называл патологию «болезнью рыбьей чешуи». Нюансы, которые легли в основу дифференцировки клинических проявлений ихтиоза, принадлежат, в том числе, Л.
Врожденная ихтиозиформная эритродермия Брока
Причины
Причины возникновения врожденной ихтиозиформной эритродермии Брока связаны с наследственной, генной патологией, обусловленной различным уровнем поражения генетического аппарата и клинически проявляющейся в разных составляющих дермы. При врождённой ихтиозиформной эритродермии Брока страдает, прежде всего, эпидермис. Его толщина, работа железистого аппарата кожи, пигментация определяются особенностями генотипа человека. Гены контролируют выработку белков и активность ферментных процессов. «Некорректная» работа ферментов отражается в клинике заболевания.
Состояние кожи, при котором превалирует образование сухих, мертвых клеток дермы на отдельных участках кожного покрова или по всей её поверхности, связано с типом наследования: аутосомно–доминантным (врождённая буллёзная ихтиозиформная эритродермия) или аутосомно-рецессивным (врождённая небуллёзная ихтиозиформная эритродермия). Буллезная форма обусловлена мутациями в генах, расположенных на 12 и 17 хромосомах и кодирующих синтез кератина 1 и 10; небуллезная форма возникает при мутации гена эпидермальной трансглутаминазы, локализованного на хромосоме 14q11. От типа наследуемых мутаций зависит тяжесть клинических проявлений и прогноз возникшего заболевания. Доминантный тип наследования характеризуется более мягким течением и благоприятным прогнозом, рецессивный – наоборот. Закономерности, приводящие к мутациям, изучены не до конца. Кровное родство родителей повышает риск заболевания у детей.
Буллезная форма обусловлена мутациями в генах, расположенных на 12 и 17 хромосомах и кодирующих синтез кератина 1 и 10; небуллезная форма возникает при мутации гена эпидермальной трансглутаминазы, локализованного на хромосоме 14q11. От типа наследуемых мутаций зависит тяжесть клинических проявлений и прогноз возникшего заболевания. Доминантный тип наследования характеризуется более мягким течением и благоприятным прогнозом, рецессивный – наоборот. Закономерности, приводящие к мутациям, изучены не до конца. Кровное родство родителей повышает риск заболевания у детей.
Аутосомно-доминантный тип наследования всегда даёт 50% здоровых наследников, 50% больных. При аутосомно-рецессивном типе наследования дерматоза возможны варианты. Если оба родителя не больны, но являются носителями патологии, то в 25% их дети заболеют обязательно; 25% будут здоровы, а 50% унаследуют носительство. Если один из родителей болен, а второй – носитель, то вероятность заболевания составляет 50%. Если больны оба родителя, вариантов здорового рождения нет. Если у здоровых родителей рождается больной ребёнок — это новая спонтанная мутация гена.
Если больны оба родителя, вариантов здорового рождения нет. Если у здоровых родителей рождается больной ребёнок — это новая спонтанная мутация гена.
Симптомы
Клинически врождённая ихтиозиформная эритродермия Брока делится на два варианта: буллёзный (с образованием пузырей) и небуллёзный («сухой»). Небуллёзная врождённая эритродермия Брока характеризуется яркой диффузной врождённой эритемой, плотной на ощупь, на фоне которой появляется обильное шелушение в виде гиперкератотических, полупрозрачных коричневых чешуек, способных отделяться целыми пластами. Кроме сухости, присутствует ощущение стянутости кожи.
При лёгкой степени дерматоза в процесс вовлекаются только лицо, ладони и подошвы. Тяжёлое течение захватывает тотально весь кожный покров. Ребёнок не может нормально дышать, сосать, открывать рот. Потовые железы работают плохо, поэтому жара или малейшая физическая нагрузка приводят к подъёму температуры до 40 градусов. Через трещины в эпидермисе теряется влага, наступает обезвоживание. Часто такие изменения заканчиваются летальным исходом. Если малыш выживает, то кожная патология сохраняется на всю жизнь. Со временем к кожным проявлениям присоединяется выпадение волос, вплоть до полного облысения; нарушается работа потовых апокриновых желёз. Кожа становится плотной и в области губ, ушей, век приводит к деформациям (выворотам кожи).
Часто такие изменения заканчиваются летальным исходом. Если малыш выживает, то кожная патология сохраняется на всю жизнь. Со временем к кожным проявлениям присоединяется выпадение волос, вплоть до полного облысения; нарушается работа потовых апокриновых желёз. Кожа становится плотной и в области губ, ушей, век приводит к деформациям (выворотам кожи).
Отличительной особенностью врождённой буллёзной ихтиозиформной эритродермии служит появление пузырей и элементов отслоения эпидермиса на фоне эритематозно изменённой кожи сразу же после рождения. Кожный покров отёчен, утолщён. Легкая степень заболевания характеризуется единичными первичными элементами с роговыми чешуйками, самостоятельным разрешением процесса к 3-5 годам. При тяжелой степени пузыри покрывают весь кожный покров, вскрываются, образуя участки мокнутия, создавая риск присоединения вторичной инфекции, трансформируются в гиперкератотические корки.
Если процесс локализуется в складках кожи, на разгибательных локтевых поверхностях, в подколенных ямках, подмышечных впадинах образуются бородавчатые разрастания мягкой консистенции, прочно прилегающие к дерме, образующие рисунок, напоминающий вельвет.
Диагностика
Диагностируют заболевание на основании анамнеза, клиники и генетического тестирования. Гистологически при «сухом» варианте зернистый слой эпидермиса гипертрофирован, в дерме определяется воспалительный инфильтрат, акантоз. Пузырный вариант даёт зернистую дегенерацию клеток мальпигиева слоя. Дифференцируют врожденную ихтиозиформную эритродермию Брока со всеми разновидностями истинного ихтиоза на основании гистологического исследования. Дополнительно проводят бакпосев крови, отделяемого пузырей с целью исключения сепсиса, вторичной инфекции.
Лечение врожденной ихтиозиформной эритродермии Брока
Патогенетическое лечение отсутствует. С учётом тяжести процесса новорожденных помещают в реанимационное отделение, поддерживая высокую влажность воздуха и контролируя обезвоживание, признаки вторичной инфекции. В первые дни проводят консультации дерматолога и генетика. Показаны ванны с последующим увлажнением кожи смягчающими кремами (детский крем). Ударное средство основной терапии – ретиноиды (витамин А, его синтетические аналоги). Принимают их курсами, по индивидуальным схемам, в больших дозировках. Периодически проводят полное клинико-диагностическое обследование с целью мониторинга функционального состояния печени, почек, сердечной мышцы. При буллёзной форме отмечена резистентность к препаратам группы витамина А, в этом случае к лечению подключают кортикостероиды. Рекомендована витаминотерапия. Хороший эффект дают ванны с отрубями, крахмалом, морской солью. Наружно применяют смягчающие, кортикостероидные мази с добавлением 3% салициловой кислоты в пропорции 1:1.
С учётом тяжести процесса новорожденных помещают в реанимационное отделение, поддерживая высокую влажность воздуха и контролируя обезвоживание, признаки вторичной инфекции. В первые дни проводят консультации дерматолога и генетика. Показаны ванны с последующим увлажнением кожи смягчающими кремами (детский крем). Ударное средство основной терапии – ретиноиды (витамин А, его синтетические аналоги). Принимают их курсами, по индивидуальным схемам, в больших дозировках. Периодически проводят полное клинико-диагностическое обследование с целью мониторинга функционального состояния печени, почек, сердечной мышцы. При буллёзной форме отмечена резистентность к препаратам группы витамина А, в этом случае к лечению подключают кортикостероиды. Рекомендована витаминотерапия. Хороший эффект дают ванны с отрубями, крахмалом, морской солью. Наружно применяют смягчающие, кортикостероидные мази с добавлением 3% салициловой кислоты в пропорции 1:1.
Прогноз и профилактика
Жить с кожей, поражённой врождённой ихтиозиформной эритродермией Брока сложно, особенно детям. Иногда больным требуется поддержка психолога. Необходимо постоянное диспансерное наблюдение за пациентом с ежегодной противорецидивной терапией. В периоды ремиссии – ежедневное, длительное применение смягчающих кожу кремов. Прогноз зависит от тяжести патологии, своевременности начатого лечения. Возможен летальный исход.
Иногда больным требуется поддержка психолога. Необходимо постоянное диспансерное наблюдение за пациентом с ежегодной противорецидивной терапией. В периоды ремиссии – ежедневное, длительное применение смягчающих кожу кремов. Прогноз зависит от тяжести патологии, своевременности начатого лечения. Возможен летальный исход.
Поскольку врожденная ихтиозиформная эритродермия Брока – это редкий генный дерматоз, с профилактической целью при планировании беременности необходимо пренатальное медико-генетическое обследование супружеских пар с отягощённым наследственным анамнезом, последующая консультация генетика, принятие взвешенного решения о деторождении. При выявлении патологии в процессе беременности показано её прерывание.
Вы можете поделиться своей историей болезни, что Вам помогло при лечении врожденной ихтиозиформной эритродермии Брока.
Источники
- Настоящая статья подготовлена по материалам сайта: https://www.krasotaimedicina.
 ru/
ru/
 ru/
ru/ВАЖНО
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Эритродермия врождённая ихтиозная (небуллёзная), TGM1 м.
Метод определения Секвенирование. Выдаётся заключение врача-генетика!
Исследуемый материал Цельная кровь (с ЭДТА)
Доступен выезд на дом
Эритродермия врожденная ихтиозная (небуллезная, OMIM242100) или аутосомно-рецессивный врожденный ихтиоз — клинически и генетически гетерогенная группа заболеваний кератинизации. Эти заболевания характеризующаяся шелушением кожи на всём теле, часто ассоциированном с эритемой. Существуют две несиндромальные формы ихтиоза: ламеллярный ихтиоз (lamellar ichthyosis — LI) и небуллёзная врождённая эритродермия (Nonbullous congenital ichthyosiform erythroderma, NCIE, MIM 242100). Частота обеих форм заболевания 1:300-500 тысяч новорождённых.
Частота обеих форм заболевания 1:300-500 тысяч новорождённых.
Клинически LI отличается от NCIE характеристикой чешуек (они крупные, тёмные) и отсутствием эритемы. При NCIE чешуйки тонкие, серебристые, располагаются на фоне эритемы, на конечностях они больше и темнее. Существуют перекрывающиеся фенотипы. При NCIE больные имеют ладонно-подошвенный кератоз, часто с болезненными трещинами, контрактурами. У половины больных встречается дистрофия ногтей. При NCIE чаще, чем при LI встречается эктропион, вовлечение кожи головы, потеря бровей и ресниц.
Тип наследования.
Аутосомно- рецессивный.
Гены, ответственные за развитие заболевания.
Ген TGM1 (keratinocyte transglutaminase 1) расположен на хромосоме 14 в регионе 14q12. Содержит 15 экзонов.
При врожденной ихтиозиформной эритродермии мутации в гене TGM1 встречаются в 38% случаев, в гене ALOXE3 – в 6,8% случаев и при мутациях в гене ALOX12B – в 6,8% случаев.
Патогенез и клиническая картина.
В основе патологического процесса в коже лежат нарушения ороговения, проявляющиеся избыточной продукцией кератина, обычно с изменением его структуры или замедлением процесса отторжения ороговевших клеток с поверхности кожи из-за цементирующего воздействия накоплений гликозаминогликанов в эпидермисе. Кодируемый геном ALOXE3фермент катализируют оксигенацию свободных и эстерификацию полиненасыщенных жирных кислот для образования соответствующих гидродериватов.
Кодируемый геном ALOXE3фермент катализируют оксигенацию свободных и эстерификацию полиненасыщенных жирных кислот для образования соответствующих гидродериватов.
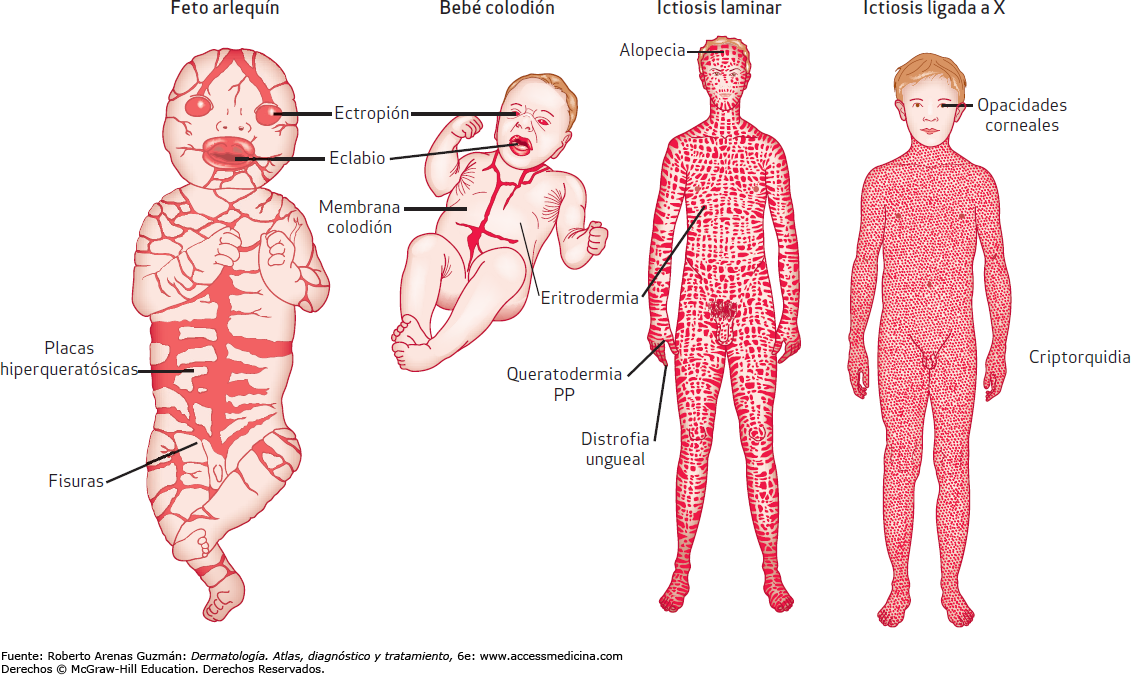
Поражение кожи появляется на 4—5-м месяце внутриутробного развития. Кожа новорожденного покрыта панцирем, состоящим из толстых роговых щитков серо-черного цвета толщиной до 10 мм, гладких и зазубренных, разделенных бороздами и трещинами. Губы ребенка малоподвижны, ротовое отверстие растянуто или резко сужено (едва проходит зонд). Нос и ушные раковины деформированы, заполнены роговыми наслоениями; веки выворочены, волосы и ногти могут отсутствовать; отмечаются пороки развития конечностей — косорукость, косолапость, контрактуры, межпальцевые перемычки.
Вся кожа ребенка с момента рождения покрыта тонкой сухой желтовато-коричневого цвета пленкой, напоминающей коллодий («коллоидный» плод). Клинически при данной форме заболевания чешуйки тонкие, серебристые, располагаются на фоне эритемы, на конечностях они больше и темнее. При данной форме, как правило, отсутствует ладонно-подошвенный кератоз, У половины больных встречается дистрофия ногтей. Возможен эктропион.
При данной форме, как правило, отсутствует ладонно-подошвенный кератоз, У половины больных встречается дистрофия ногтей. Возможен эктропион.
Эритродермия слабо выражена. Проявления ихтиоза обычно выражены мягче, чем при мутациях в гене ALOX12B.
В ряде случаев чешуйки исчезают в грудном возрасте, в дальнейшем кожа на протяжении всей жизни остается нормальной.
Частота встречаемости: 1:300-500 000 новорожденных.
Эпидермолитический гиперкератоз — StatPearls — NCBI Bookshelf
Continuing Education Activity
Эпидермолитический гиперкератоз (буллезная ихтиозиформная эритродермия) — редкое аутосомно-доминантное заболевание ороговения, вызванное мутациями в кератинах 1 и 10. Оно характеризуется эритродермией и образованием пузырей при рождении с последующее развитие гиперкератоза. В этом мероприятии рассматривается оценка и лечение эпидермолитического гиперкератоза и объясняется роль межпрофессиональной команды в улучшении ухода за пациентами с этим заболеванием.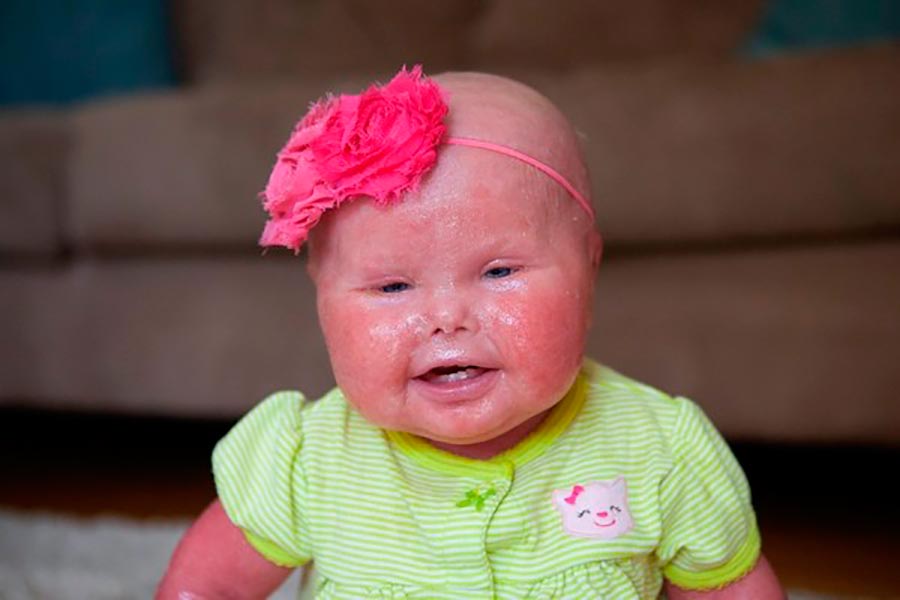
Цели:
Определите этиологию эпидермолитического гиперкератоза (буллезная ихтиозиформная эритродермия).
Изучите анамнез, физикальное обследование и оценку эпидермолитического гиперкератоза (буллезная ихтиозиформная эритродермия).
Опишите возможные варианты лечения эпидермолитического гиперкератоза (буллезная ихтиозиформная эритродермия).
Опишите стратегии межпрофессиональной бригады по улучшению координации помощи и коммуникации для прогрессирования эпидермолитического гиперкератоза (буллезной ихтиозиформной эритродермии) и улучшения исходов.
Получите доступ к бесплатным вопросам с несколькими вариантами ответов по этой теме.
. Введение. выраженный гиперкератоз. Это проявление происходит с ладонно-подошвенной кератодермией или без нее. Эпидермолитический гиперкератоз легко отличить от других форм врожденного ихтиоза по характерным гистологическим признакам.
 В более поздней литературе эпидермолитический гиперкератоз имеет обозначение как патологический термин, а заболевание носит название эпидермолитического ихтиоза.[1][2][3]
В более поздней литературе эпидермолитический гиперкератоз имеет обозначение как патологический термин, а заболевание носит название эпидермолитического ихтиоза.[1][2][3]Этиология
Эпидермолитический гиперкератоз в первую очередь является результатом точечных или миссенс-мутаций в генах, кодирующих кератин 1 (KRT1) и кератин 10 (KRT10).[1][4][2][5][6] В большинстве случаев передается по аутосомно-доминантному типу наследования.[1][2][7] Тяжелая форма эпидермолитического гиперкератоза существует у трех кровнородственных родственников из-за аутосомно-рецессивных мутаций потери функции в кератине 10.[2][8][9] Спорадические мутации встречаются до 50% случаев.[1][2][7] Мутации в кератине 1 были связаны с эпидермолитическим гиперкератозом с тяжелой ладонно-подошвенной кератодермией, в то время как мутации в кератине 10 вызывают фенотип без ладонно-подошвенной кератодермии.
У потомства родителей с эпидермальными невусами может быть генерализованный эпидермолитический гиперкератоз из-за генетического мозаицизма. Мозаичная форма эпидермолитического гиперкератоза, по-разному обозначаемая как ichthyosis hystrix или линейный эпидермолитический гиперкератоз, вызывается постзиготными мутациями в кератине 1 и кератине 10 во время эмбриогенеза. Если эти мутации затрагивают клетки гонад, они могут передаваться потомству и вызывать фенотипический эпидермолитический гиперкератоз.[7][11] В настоящее время эпидермолитический гиперкератоз является единственным известным кератиновым заболеванием человека, проявляющим генетический мозаицизм.[7]
Мозаичная форма эпидермолитического гиперкератоза, по-разному обозначаемая как ichthyosis hystrix или линейный эпидермолитический гиперкератоз, вызывается постзиготными мутациями в кератине 1 и кератине 10 во время эмбриогенеза. Если эти мутации затрагивают клетки гонад, они могут передаваться потомству и вызывать фенотипический эпидермолитический гиперкератоз.[7][11] В настоящее время эпидермолитический гиперкератоз является единственным известным кератиновым заболеванием человека, проявляющим генетический мозаицизм.[7]
Эпидемиология
Сообщалось, что эпидермолитический гиперкератоз поражает от 1 из 200 000 до 300 000 младенцев.[1][12] Расстройство является преимущественно аутосомно-доминантным, и, следовательно, нет гендерной предрасположенности.[1]
Патофизиология
Эпидермолитический гиперкератоз вызывается мутациями, разрушающими кератиновую сеть эпидермиса. Белки кератина являются наиболее важными белками, необходимыми для структурного развития эпидермиса.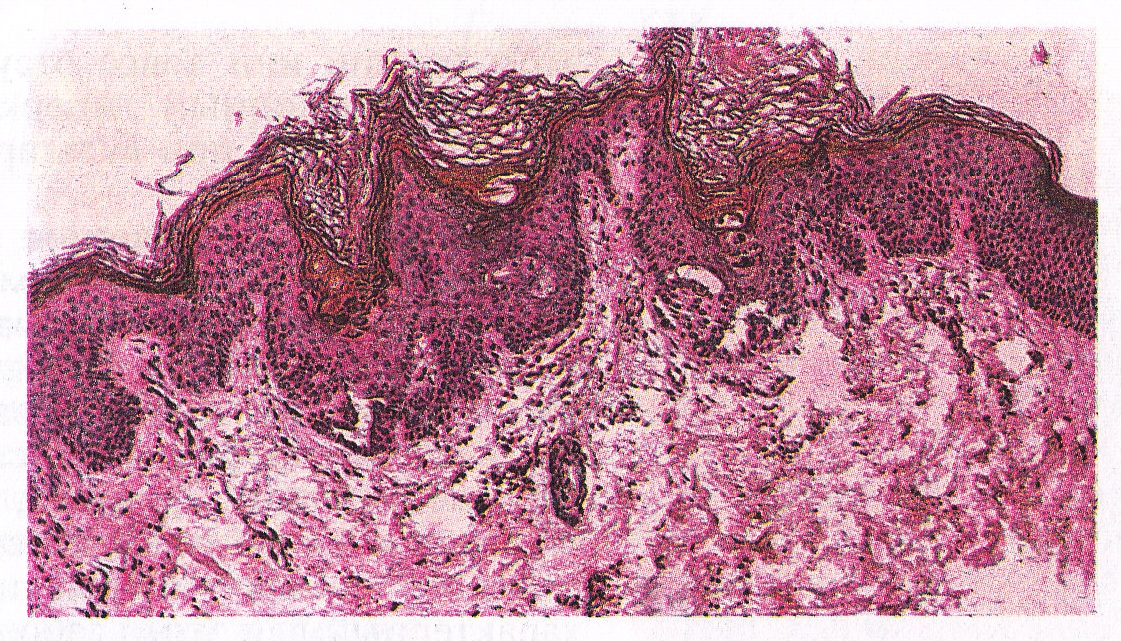 [1][13] Эпидермис защищает кожу от условий окружающей среды, образуя массивную цитоскелетную структуру из кератиновых филаментов. Гены, кодирующие кератин 5 и кератин 14, экспрессируются в базальном слое эпидермиса. Поскольку эпидермальные клетки продолжают окончательно дифференцироваться, супрабазальные клетки состоят из кератиновых 1 и кератиновых 10 филаментов, которые толще, чем пучки тонофиламентов базальных клеток. Более толстые пучки тонофиламентов обеспечивают повышенную выживаемость кератинов.[1][14][15]
[1][13] Эпидермис защищает кожу от условий окружающей среды, образуя массивную цитоскелетную структуру из кератиновых филаментов. Гены, кодирующие кератин 5 и кератин 14, экспрессируются в базальном слое эпидермиса. Поскольку эпидермальные клетки продолжают окончательно дифференцироваться, супрабазальные клетки состоят из кератиновых 1 и кератиновых 10 филаментов, которые толще, чем пучки тонофиламентов базальных клеток. Более толстые пучки тонофиламентов обеспечивают повышенную выживаемость кератинов.[1][14][15]
Мутации при эпидермолитическом гиперкератозе происходят в высококонсервативных а-спиральных стержневых доменах кератина 1 и кератина 10. Мутации в мотивах пограничной последовательности спирали влияют на мотивы инициации и терминации спирали и приводят к значительному нарушению сборки филаментов, агрегации тонофиламентов и, как правило, к более тяжелым фенотипы. Скомпрометированный эпидермис, в свою очередь, склонен к цитолизу и образованию пузырей, что приводит к нарушению барьерной функции кожи, вызывая повышенную трансэпидермальную потерю воды и бактериальную колонизацию. [7] Гиперкератоз возникает в результате гиперпролиферации базальных клеток и уменьшения шелушения [1].
[7] Гиперкератоз возникает в результате гиперпролиферации базальных клеток и уменьшения шелушения [1].
Чипев и др. с помощью электронной микроскопии обнаружили точечную мутацию лейцина в пролин в субдомене h2 промежуточного кератинового филамента 1, которая значительно нарушила структуру и организацию кератиновых филаментов [4]. Снайдер и др. идентифицировали мутацию аргинина в гистидин на амино-конце а-спирального стержневого домена KRT10 и мутацию тирозина в цистеин в карбокси-домене KRT1. Тяжелые формы эпидермолитического гиперкератоза связаны с делецией экзона 6 гена KRT1[6] и миссенс-мутациями (c.475T>C;p.Ser159).Pro и c.562A>C;p.N188H) в KRT10 и KRT1 соответственно.[1][17]
Гистопатология
Характерные гистопатологические данные отличают эпидермолитический гиперкератоз от других врожденных ихтиозов. Выдающиеся особенности гистологии включают плотный ортогиперкератоз, гипергранулез, грубые кератогиалиновые гранулы и цитолиз супрабазального и зернистого слоев. Кератиноциты демонстрируют выраженную внутриклеточную вакуолярную дегенерацию и плотные скопления кератиновых промежуточных филаментов. Также встречаются дискератоз различной степени и легкая периваскулярная лимфогистиоцитарная инфильтрация. Электронная микроскопия показывает слипшиеся кератиновые промежуточные филаменты в супрабазальном слое, а иммуногистохимия показывает дефекты кератинов 1 или 10.[12] У пациентов с мозаичной формой эпидермолитического гиперкератоза обнаруживаются фокальные очаги поражения с пропуском участков нормального эпидермиса.[1][2]
Кератиноциты демонстрируют выраженную внутриклеточную вакуолярную дегенерацию и плотные скопления кератиновых промежуточных филаментов. Также встречаются дискератоз различной степени и легкая периваскулярная лимфогистиоцитарная инфильтрация. Электронная микроскопия показывает слипшиеся кератиновые промежуточные филаменты в супрабазальном слое, а иммуногистохимия показывает дефекты кератинов 1 или 10.[12] У пациентов с мозаичной формой эпидермолитического гиперкератоза обнаруживаются фокальные очаги поражения с пропуском участков нормального эпидермиса.[1][2]
Анамнез и физикальное исследование
Эпидермолитический гиперкератоз появляется при рождении с генерализованной эритродермией. Хрупкость кожи вызывает волдыри, шелушение/шелушение, эрозии и обширные участки оголенной кожи даже при незначительной травме. Через несколько месяцев эритема и образование пузырей уменьшаются, развивается выраженный гиперкератоз. Иногда сохраняется ломкость кожи, и пациенты периодически отслаивают большие пластины поверхностного эпидермиса. Гиперкератоз характерно напоминает «гофрированный картон» при наложении на области сгибов или «булыжную мостовую» при наложении на разгибательные поверхности суставов. Иногда происходит тяжелое поражение кожи головы и шеи, что приводит к инкапсуляции волосяных стержней и алопеции. Бактериальная колонизация мацерированных чешуек вызывает характерный неприятный запах, напоминающий запах тухлых яиц. Обычно сопутствующие симптомы включают ксероз, зуд, болезненные трещины, ангидроз и уменьшение объема движений в суставах.[1][12]
Гиперкератоз характерно напоминает «гофрированный картон» при наложении на области сгибов или «булыжную мостовую» при наложении на разгибательные поверхности суставов. Иногда происходит тяжелое поражение кожи головы и шеи, что приводит к инкапсуляции волосяных стержней и алопеции. Бактериальная колонизация мацерированных чешуек вызывает характерный неприятный запах, напоминающий запах тухлых яиц. Обычно сопутствующие симптомы включают ксероз, зуд, болезненные трещины, ангидроз и уменьшение объема движений в суставах.[1][12]
Исследователи выявили несколько различных фенотипов эпидермолитического гиперкератоза. В 1994 году DiGiovanna и Bale описали две основные клинические категории: с ладонно-подошвенной кератодермией и без ладонно-подошвенной кератодермии [18]. Каждая клиническая категория имеет три подтипа с различной степенью эритродермии, образованием пузырей, шелушением (неладонно-подошвенный тип) и поражением туловища (ладонно-подошвенный тип) [12]. Контрактуры пальцев из-за ладонно-подошвенного поражения могут привести к функциональным нарушениям. Мозаичная форма эпидермолитического гиперкератоза характеризуется односторонними или двусторонними полосами гиперкератоза по Блашкоидному типу.[7][11] Те, у кого более обширное поражение и поражения с выступающими, похожими на дикобраза шипами, были названы «ихтиозом гистрикса».0005
Мозаичная форма эпидермолитического гиперкератоза характеризуется односторонними или двусторонними полосами гиперкератоза по Блашкоидному типу.[7][11] Те, у кого более обширное поражение и поражения с выступающими, похожими на дикобраза шипами, были названы «ихтиозом гистрикса».0005
Оценка
Основой диагностики являются клинические, гистопатологические и лабораторные данные. Анализ генетических мутаций дефектов кератина с помощью мультигенного скрининга в настоящее время является золотым стандартом и может использоваться для пренатального скрининга.[1][12] Пренатальную диагностику можно получить с помощью забора ворсин хориона, амниоцентеза и фетоскопии с биопсией кожи плода. Однако эти исследования имеют ограничения низкой чувствительности или генетической гетерогенности. Оптические и спектроскопические методы, такие как рамановская спектроскопия и оптическая когерентная томография, использовались для идентификации немеланомного рака кожи in vivo и могут быть потенциальным методом диагностики эпидермолитического гиперкератоза.
Лечение/управление
Лечение преимущественно симптоматическое и зависит от возраста пациента и клинических проявлений. Младенцы должны находиться под наблюдением в условиях отделения интенсивной терапии для лечения обезвоживания, электролитного дисбаланса и кожной суперинфекции. Лечение сепсиса должно включать внутривенное введение антибиотиков широкого спектра действия. Для защиты кожи и заживления оголенных участков следует использовать местные смягчающие средства и защитные прокладки.[12]
Целью терапии у детей и взрослых является уменьшение гиперкератоза. Местные смягчающие и кератолитические средства, содержащие глицерин, молочную кислоту, мочевину и а-гидроксикислоты, продемонстрировали улучшение при гиперкератозе, но часто плохо переносятся из-за жжения и покалывания. Клиницистам следует избегать широкого применения препаратов для местного применения с более высокой концентрацией салициловой кислоты из-за риска системного салицилизма.[1][12] Было показано, что местные ретиноиды, N-ацетилцистеин, лиарозол и кальципотриол влияют на функцию корнеоцитов и снижают гиперпролиферацию эпидермиса.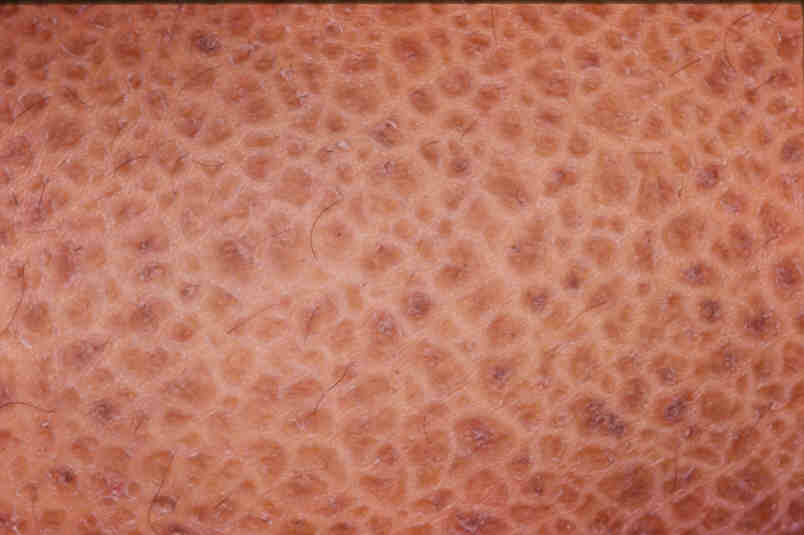 Однако эти агенты также могут вызывать раздражение кожи.[1] В тяжелых случаях эпидермолитического гиперкератоза пероральные ретиноиды могут значительно уменьшить гиперкератоз и частоту суперинфекций, но могут парадоксальным образом повысить ломкость кожи и усугубить образование волдырей. Таким образом, рекомендуется использовать низкие начальные дозы, а затем постепенно увеличивать их под тщательным контролем [19].] Следует отметить, что пациенты с мутациями кератина 10 лучше реагируют на местные или системные ретиноиды по сравнению с пациентами с мутациями кератина 1.[1][12] Антибактериальные мыла, хлоргексидин и ванны с разбавленным гипохлоритом натрия могут уменьшить бактериальную колонизацию. При возникновении бактериальной инфекции кожи необходимы местные или системные антибиотики.[1][12]
Однако эти агенты также могут вызывать раздражение кожи.[1] В тяжелых случаях эпидермолитического гиперкератоза пероральные ретиноиды могут значительно уменьшить гиперкератоз и частоту суперинфекций, но могут парадоксальным образом повысить ломкость кожи и усугубить образование волдырей. Таким образом, рекомендуется использовать низкие начальные дозы, а затем постепенно увеличивать их под тщательным контролем [19].] Следует отметить, что пациенты с мутациями кератина 10 лучше реагируют на местные или системные ретиноиды по сравнению с пациентами с мутациями кератина 1.[1][12] Антибактериальные мыла, хлоргексидин и ванны с разбавленным гипохлоритом натрия могут уменьшить бактериальную колонизацию. При возникновении бактериальной инфекции кожи необходимы местные или системные антибиотики.[1][12]
Дифференциальный диагноз
Дифференциальный диагноз эпидермолитического гиперкератоза включает другие причины эритродермии, булл/волдырей или шелушения в детском возрасте.
Другие врожденные ихтиозы:
Поверхностный эпидермолитический ихтиоз
Ламеллярный ихтиоз
Врожденный ихтиозиформный 0027
Везикулобуллезные и эрозивные заболевания у детей:
Буллезный эпидермолиз
Стафилококковый ошпаренный кожный покров синдром
Буллезное импетиго
Простой герпес
Врожденный эрозивно-везикулярный дерматоз
Аутоиммунные пузырчатые болезни
Генодерматозы:
4 Синдром Шегрена-Ларссона 11
Болезнь накопления нейтральных липидов
Трихотиодистрофия
Синдром Нетертона
Дефицит стероидной сульфатазы
Синдром шелушения кожи
Синдром Конради-Хунерманна-Гаппла
ДЕТСКИЙ синдром
ДЕТСКИЙ синдром
Прогноз
Тяжесть эпидермолитического гиперкератоза варьирует.
Новорожденные с этим заболеванием подвергаются повышенному риску сепсиса, обезвоживания и, в конечном итоге, смерти. Из-за редкости этого заболевания статистические данные о смертности отсутствуют. У тех, кто пережил неонатальный период, эпизоды образования волдырей и кожных инфекций периодически возникают на протяжении всей жизни. В результате у пациентов часто наблюдается значительное обезображивание и резкий запах тела, что вызывает серьезные психологические и социальные расстройства.[1][12]Осложнения
Новорожденные, рожденные с эпидермолитическим гиперкератозом, подвержены более высокому риску обезвоживания, дисбаланса электролитов и сепсиса из-за повышенной трансэпидермальной потери воды и неэффективной барьерной функции кожи. Без надлежащего лечения эти осложнения приводят к летальному исходу. Кроме того, серьезное ладонно-подошвенное поражение может привести к контрактурам пальцев, что негативно влияет на подвижность суставов, походку и осанку.[12]
Сдерживание и обучение пациентов
У пациентов с эпидермолитическим гиперкератозом на протяжении всей жизни наблюдается повышенная хрупкость кожи, поэтому важно обучать пациентов способам сведения к минимуму механической травмы.
Это возможно, если носить свободную, удобную одежду и хорошо сидящую обувь.Улучшение результатов медицинского персонала
Эпидермолитический гиперкератоз обычно проявляется вскоре после рождения. Это неизлечимо, и расстройство прогрессирует. Лечение в первую очередь симптоматическое и зависит от возраста пациента и клинической картины. Врачебное ведение включает семейного врача, педиатра и дерматолога, а также НП и ПА. Младенцы должны находиться под наблюдением в условиях отделения интенсивной терапии для лечения обезвоживания, электролитного дисбаланса и кожной суперинфекции. Сепсис следует лечить внутривенными антибиотиками широкого спектра действия, которые должны быть согласованы с фармацевтом в отношении охвата, дозирования и мониторинга лекарственных взаимодействий. Медсестра должна обучить опекуна или родителя использованию местных смягчающих средств и защитных прокладок для защиты кожи и заживления оголенных участков, которые также могут быть использованы в аптеке.
Поскольку кожа хрупкая, практикующая медсестра должна также информировать пациентов о том, как свести к минимуму механическую травму, что достижимо при ношении свободной, удобной одежды и удобной обуви.Из-за значительной психосоциальной заболеваемости, вызванной отдаленными последствиями эпидермолитического гиперкератоза, необходимо координировать лечение с помощью межпрофессиональной бригады, включающей реаниматологов, инфекционистов, психиатрических медсестер и психиатров. Задержки в диагностике и лечении могут привести к негативным последствиям и возможной смерти. Все эти члены межпрофессиональной медицинской бригады должны сотрудничать и общаться, чтобы добиться положительных результатов лечения пациентов.[1] [Уровень 5]
Контрольные вопросы
Доступ к бесплатным вопросам с несколькими вариантами ответов по этой теме.
Комментарий к этой статье.
Рисунок
Эпидермолитический гиперкератоз. Изображение предоставлено S Bhimji MD
Рисунок
Эпидермолитический гиперкератоз.
Предоставлено д-ром Shyam Verma, MBBS, DVD, FRCP, FAAD, Vadodara, IndiaСсылки
- 1.
Peter Rout D, Nair A, Gupta A, Kumar P. Эпидермолитический гиперкератоз: клиническое обновление. Clin Cosmet Investig Dermatol. 2019;12:333-344. [Статья бесплатно PMC: PMC6512611] [PubMed: 31190940]
- 2.
Covaciu C, Castori M, De Luca N, Ghirri P, Nannipieri A, Ragone G, Zambruno G, Castiglia D. Летальный аутосомно-рецессивный эпидермолитический эпидермолитический из-за новой мутации донорского сайта сплайсинга в KRT10. Бр Дж Дерматол. 2010 июнь; 162 (6): 1384-7. [PubMed: 20302579]
- 3.
Оджи В., Тадини Г., Акияма М., Бланше Бардон С., Бодемер С., Бурра Э., Кудьер П., ДиДжиованна Дж.Дж., Элиас П., Фишер Дж., Флекман П., Джина М., Харпер Дж., Хашимото Т., Хауссер И., Хеннис Х.К., Холь Д., Овнанян А., Исида-Ямамото А., Яцик В.К., Личман С., Ли И., Мазереу-Хотье Дж., Милстон Л., Морис-Пикард Ф., Паллер А.С., Ричард Г.
, Schmuth M, Shimizu H, Sprecher E, Van Steensel M, Taïeb A, Toro JR, Vabres P, Vahlquist A, Williams M, Traupe H. Пересмотренная номенклатура и классификация наследственных ихтиозов: результаты Первой консенсусной конференции по ихтиозу в Сорезе, 2009 г.. J Am Acad Дерматол. 2010 Октябрь; 63 (4): 607-41. [PubMed: 20643494]- 4.
Чипев С.С., Корге Б.П., Маркова Н., Бейл С.Дж., ДиДжиованна Д.Дж., Комптон Д.Г., Штайнерт П.М. Мутация лейцина—-пролина в субдомене h2 кератина 1 вызывает эпидермолитический гиперкератоз. Клетка. 1992 Сентябрь 04; 70 (5): 821-8. [PubMed: 1381288]
- 5.
Исида-Ямамото А., МакГрат Дж.А., судья М.Р., Ли И.М., Лейн Э.Б., Иди Р.А. Избирательное участие кератинов К1 и К10 в цитоскелетной аномалии эпидермолитического гиперкератоза (буллезная врожденная ихтиозиформная эритродермия). Джей Инвест Дерматол. 1992 июля; 99(1):19-26. [PubMed: 1376754]
- 6.
Виртанен М., Смит С.К., Гедде-Даль Т.
, Валквист А., Боуден П.Е. Мутации сайта сплайсинга и делеции в генах кератина (KRT1 и KRT10): необычные фенотипические изменения у скандинавских пациентов с эпидермолитическим гиперкератозом. Джей Инвест Дерматол. 2003 ноябрь; 121(5):1013-20. [PubMed: 14708600]- 7.
Ирвин А.Д., Маклин В.Х. Кератиновые заболевания человека: увеличивающийся спектр заболеваний и тонкость корреляции фенотип-генотип. Бр Дж Дерматол. 1999 мая; 140(5):815-28. [PubMed: 10354017]
- 8.
Мюллер Ф.Б., Хубер М., Кинаджян Т., Хауссер И., Шаффрат С., Криг Т., Холь Д., Корге Б.П., Арин М.Дж. Нокаут кератина 10 человека вызывает рецессивный эпидермолитический гиперкератоз. Хум Мол Жене. 01 апреля 2006 г.; 15 (7): 1133-41. [PubMed: 16505000]
- 9.
Цубота А., Акияма М., Канитакис Дж., Сакаи К., Номура Т., Клауди А., Симидзу Х. Легкая рецессивная буллезная врожденная ихтиозиформная эритродермия из-за ранее невыявленной гомозиготной мутации 10 .
Джей Инвест Дерматол. 2008 июль; 128 (7): 1648-52. [В паблике: 18219278]- 10.
Ди Джованна Дж.Дж., Бэйл С.Дж. Клиническая гетерогенность эпидермолитического гиперкератоза. Арка Дерматол. 1994 г., август; 130(8):1026-35. [PubMed: 8053700]
- 11.
Paller AS, Syder AJ, Chan YM, Yu QC, Hutton E, Tadini G, Fuchs E. Генетический и клинический мозаицизм в типе эпидермального невуса. N Engl J Med. 1994 24 ноября; 331 (21): 1408-15. [PubMed: 7526210]
- 12.
Квак Дж., Маверакис Э. Эпидермолитический гиперкератоз. Dermatol Online J. 2006, 08 сентября; 12 (5): 6. [В паблике: 16962021]
- 13.
Fuchs E, Coulombe P, Cheng J, Chan YM, Hutton E, Syder A, Degenstein L, Yu QC, Letai A, Vassar R. Генетические основы простого буллезного эпидермолиза и эпидермолитического гиперкератоза. Джей Инвест Дерматол. 1994 г., ноябрь; 103 (5 доп.): 25S-30S. [PubMed: 7525738]
- 14.
Nelson WG, Sun TT.
Классы кератина 50 и 58 кДа как молекулярные маркеры многослойного плоского эпителия: исследования клеточных культур. Джей Селл Биол. 1983 июль; 97(1):244-51. [Бесплатная статья PMC: PMC2112495] [PubMed: 6190820]- 15.
Roop DR, Huitfeldt H, Kilkenny A, Yuspa SH. Регулируемая экспрессия кератинов, связанных с дифференцировкой, в культивируемых клетках эпидермиса, обнаруженная моноспецифическими антителами к уникальным пептидам эпидермальных кератинов мыши. Дифференциация. 1987;35(2):143-50. [PubMed: 2450799]
- 16.
Syder AJ, Yu QC, Paller AS, Giudice G, Pearson R, Fuchs E. Генетические мутации в генах K1 и K10 у пациентов с эпидермолитическим гиперкератозом. Взаимосвязь между локализацией и тяжестью заболевания. Джей Клин Инвест. 1994 апреля; 93 (4): 1533-42. [Бесплатная статья PMC: PMC294170] [PubMed: 7512983]
- 17.
Chen PJ, Li CX, Wen J, Peng YS, Zeng K, Zhang SQ, Tian X, Zhang XB. Мутация S159P гена кератина 10 вызывает тяжелую форму эпидермолитического гиперкератоза.
J Eur Acad Dermatol Venereol. 2016 Окт;30(10):e102-e104. [PubMed: 26373619]- 18.
DiGiovanna JJ, Bale SJ. Эпидермолитический гиперкератоз: прикладная молекулярная генетика. Джей Инвест Дерматол. 1994 март; 102 (3): 390-4. [PubMed: 7509838]
- 19.
Лакур М., Мехта-Нихар Б., Атертон Д.Дж., Харпер Дж.И. Оценка терапии ацитретином у детей с наследственными нарушениями кератинизации. Бр Дж Дерматол. 1996 г., июнь; 134 (6): 1023-9. [PubMed: 8763418]
Эпидермолитический гиперкератоз — Статупидлс — NCBI Книжная полка
Непрерывное образование. эритродермией и образованием пузырей при рождении с последующим развитием гиперкератоза. В этом мероприятии рассматривается оценка и лечение эпидермолитического гиперкератоза и объясняется роль межпрофессиональной команды в улучшении ухода за пациентами с этим заболеванием.
Цели:
Определите этиологию эпидермолитического гиперкератоза (буллезная ихтиозиформная эритродермия).
Изучите анамнез, физикальное обследование и оценку эпидермолитического гиперкератоза (буллезная ихтиозиформная эритродермия).
Опишите возможные варианты лечения эпидермолитического гиперкератоза (буллезная ихтиозиформная эритродермия).
Опишите стратегии межпрофессиональной бригады по улучшению координации помощи и коммуникации для прогрессирования эпидермолитического гиперкератоза (буллезной ихтиозиформной эритродермии) и улучшения исходов.
Получите доступ к бесплатным вопросам с несколькими вариантами ответов по этой теме.
. Введение. выраженный гиперкератоз. Это проявление происходит с ладонно-подошвенной кератодермией или без нее. Эпидермолитический гиперкератоз легко отличить от других форм врожденного ихтиоза по характерным гистологическим признакам. В более поздней литературе эпидермолитический гиперкератоз имеет обозначение как патологический термин, а заболевание носит название эпидермолитического ихтиоза.
[1][2][3]Этиология
Эпидермолитический гиперкератоз в первую очередь является результатом точечных или миссенс-мутаций в генах, кодирующих кератин 1 (KRT1) и кератин 10 (KRT10).[1][4][2][5][6] В большинстве случаев передается по аутосомно-доминантному типу наследования.[1][2][7] Тяжелая форма эпидермолитического гиперкератоза существует у трех кровнородственных родственников из-за аутосомно-рецессивных мутаций потери функции в кератине 10.[2][8][9] Спорадические мутации встречаются до 50% случаев.[1][2][7] Мутации в кератине 1 были связаны с эпидермолитическим гиперкератозом с тяжелой ладонно-подошвенной кератодермией, в то время как мутации в кератине 10 вызывают фенотип без ладонно-подошвенной кератодермии.
У потомства родителей с эпидермальными невусами может быть генерализованный эпидермолитический гиперкератоз из-за генетического мозаицизма. Мозаичная форма эпидермолитического гиперкератоза, по-разному обозначаемая как ichthyosis hystrix или линейный эпидермолитический гиперкератоз, вызывается постзиготными мутациями в кератине 1 и кератине 10 во время эмбриогенеза.
Если эти мутации затрагивают клетки гонад, они могут передаваться потомству и вызывать фенотипический эпидермолитический гиперкератоз.[7][11] В настоящее время эпидермолитический гиперкератоз является единственным известным кератиновым заболеванием человека, проявляющим генетический мозаицизм.[7]Эпидемиология
Сообщалось, что эпидермолитический гиперкератоз поражает от 1 из 200 000 до 300 000 младенцев.[1][12] Расстройство является преимущественно аутосомно-доминантным, и, следовательно, нет гендерной предрасположенности.[1]
Патофизиология
Эпидермолитический гиперкератоз вызывается мутациями, разрушающими кератиновую сеть эпидермиса. Белки кератина являются наиболее важными белками, необходимыми для структурного развития эпидермиса.[1][13] Эпидермис защищает кожу от условий окружающей среды, образуя массивную цитоскелетную структуру из кератиновых филаментов. Гены, кодирующие кератин 5 и кератин 14, экспрессируются в базальном слое эпидермиса. Поскольку эпидермальные клетки продолжают окончательно дифференцироваться, супрабазальные клетки состоят из кератиновых 1 и кератиновых 10 филаментов, которые толще, чем пучки тонофиламентов базальных клеток.
Более толстые пучки тонофиламентов обеспечивают повышенную выживаемость кератинов.[1][14][15]Мутации при эпидермолитическом гиперкератозе происходят в высококонсервативных а-спиральных стержневых доменах кератина 1 и кератина 10. Мутации в мотивах пограничной последовательности спирали влияют на мотивы инициации и терминации спирали и приводят к значительному нарушению сборки филаментов, агрегации тонофиламентов и, как правило, к более тяжелым фенотипы. Скомпрометированный эпидермис, в свою очередь, склонен к цитолизу и образованию пузырей, что приводит к нарушению барьерной функции кожи, вызывая повышенную трансэпидермальную потерю воды и бактериальную колонизацию.[7] Гиперкератоз возникает в результате гиперпролиферации базальных клеток и уменьшения шелушения [1].
Чипев и др. с помощью электронной микроскопии обнаружили точечную мутацию лейцина в пролин в субдомене h2 промежуточного кератинового филамента 1, которая значительно нарушила структуру и организацию кератиновых филаментов [4].
Снайдер и др. идентифицировали мутацию аргинина в гистидин на амино-конце а-спирального стержневого домена KRT10 и мутацию тирозина в цистеин в карбокси-домене KRT1. Тяжелые формы эпидермолитического гиперкератоза связаны с делецией экзона 6 гена KRT1[6] и миссенс-мутациями (c.475T>C;p.Ser159).Pro и c.562A>C;p.N188H) в KRT10 и KRT1 соответственно.[1][17]Гистопатология
Характерные гистопатологические данные отличают эпидермолитический гиперкератоз от других врожденных ихтиозов. Выдающиеся особенности гистологии включают плотный ортогиперкератоз, гипергранулез, грубые кератогиалиновые гранулы и цитолиз супрабазального и зернистого слоев. Кератиноциты демонстрируют выраженную внутриклеточную вакуолярную дегенерацию и плотные скопления кератиновых промежуточных филаментов. Также встречаются дискератоз различной степени и легкая периваскулярная лимфогистиоцитарная инфильтрация. Электронная микроскопия показывает слипшиеся кератиновые промежуточные филаменты в супрабазальном слое, а иммуногистохимия показывает дефекты кератинов 1 или 10.
[12] У пациентов с мозаичной формой эпидермолитического гиперкератоза обнаруживаются фокальные очаги поражения с пропуском участков нормального эпидермиса.[1][2]Анамнез и физикальное исследование
Эпидермолитический гиперкератоз появляется при рождении с генерализованной эритродермией. Хрупкость кожи вызывает волдыри, шелушение/шелушение, эрозии и обширные участки оголенной кожи даже при незначительной травме. Через несколько месяцев эритема и образование пузырей уменьшаются, развивается выраженный гиперкератоз. Иногда сохраняется ломкость кожи, и пациенты периодически отслаивают большие пластины поверхностного эпидермиса. Гиперкератоз характерно напоминает «гофрированный картон» при наложении на области сгибов или «булыжную мостовую» при наложении на разгибательные поверхности суставов. Иногда происходит тяжелое поражение кожи головы и шеи, что приводит к инкапсуляции волосяных стержней и алопеции. Бактериальная колонизация мацерированных чешуек вызывает характерный неприятный запах, напоминающий запах тухлых яиц.
Обычно сопутствующие симптомы включают ксероз, зуд, болезненные трещины, ангидроз и уменьшение объема движений в суставах.[1][12]Исследователи выявили несколько различных фенотипов эпидермолитического гиперкератоза. В 1994 году DiGiovanna и Bale описали две основные клинические категории: с ладонно-подошвенной кератодермией и без ладонно-подошвенной кератодермии [18]. Каждая клиническая категория имеет три подтипа с различной степенью эритродермии, образованием пузырей, шелушением (неладонно-подошвенный тип) и поражением туловища (ладонно-подошвенный тип) [12]. Контрактуры пальцев из-за ладонно-подошвенного поражения могут привести к функциональным нарушениям. Мозаичная форма эпидермолитического гиперкератоза характеризуется односторонними или двусторонними полосами гиперкератоза по Блашкоидному типу.[7][11] Те, у кого более обширное поражение и поражения с выступающими, похожими на дикобраза шипами, были названы «ихтиозом гистрикса».0005
Оценка
Основой диагностики являются клинические, гистопатологические и лабораторные данные.
Анализ генетических мутаций дефектов кератина с помощью мультигенного скрининга в настоящее время является золотым стандартом и может использоваться для пренатального скрининга.[1][12] Пренатальную диагностику можно получить с помощью забора ворсин хориона, амниоцентеза и фетоскопии с биопсией кожи плода. Однако эти исследования имеют ограничения низкой чувствительности или генетической гетерогенности. Оптические и спектроскопические методы, такие как рамановская спектроскопия и оптическая когерентная томография, использовались для идентификации немеланомного рака кожи in vivo и могут быть потенциальным методом диагностики эпидермолитического гиперкератоза.Лечение/управление
Лечение преимущественно симптоматическое и зависит от возраста пациента и клинических проявлений. Младенцы должны находиться под наблюдением в условиях отделения интенсивной терапии для лечения обезвоживания, электролитного дисбаланса и кожной суперинфекции. Лечение сепсиса должно включать внутривенное введение антибиотиков широкого спектра действия.
Для защиты кожи и заживления оголенных участков следует использовать местные смягчающие средства и защитные прокладки.[12]Целью терапии у детей и взрослых является уменьшение гиперкератоза. Местные смягчающие и кератолитические средства, содержащие глицерин, молочную кислоту, мочевину и а-гидроксикислоты, продемонстрировали улучшение при гиперкератозе, но часто плохо переносятся из-за жжения и покалывания. Клиницистам следует избегать широкого применения препаратов для местного применения с более высокой концентрацией салициловой кислоты из-за риска системного салицилизма.[1][12] Было показано, что местные ретиноиды, N-ацетилцистеин, лиарозол и кальципотриол влияют на функцию корнеоцитов и снижают гиперпролиферацию эпидермиса. Однако эти агенты также могут вызывать раздражение кожи.[1] В тяжелых случаях эпидермолитического гиперкератоза пероральные ретиноиды могут значительно уменьшить гиперкератоз и частоту суперинфекций, но могут парадоксальным образом повысить ломкость кожи и усугубить образование волдырей.
Таким образом, рекомендуется использовать низкие начальные дозы, а затем постепенно увеличивать их под тщательным контролем [19].] Следует отметить, что пациенты с мутациями кератина 10 лучше реагируют на местные или системные ретиноиды по сравнению с пациентами с мутациями кератина 1.[1][12] Антибактериальные мыла, хлоргексидин и ванны с разбавленным гипохлоритом натрия могут уменьшить бактериальную колонизацию. При возникновении бактериальной инфекции кожи необходимы местные или системные антибиотики.[1][12]Дифференциальный диагноз
Дифференциальный диагноз эпидермолитического гиперкератоза включает другие причины эритродермии, булл/волдырей или шелушения в детском возрасте.
Другие врожденные ихтиозы:
Поверхностный эпидермолитический ихтиоз
Ламеллярный ихтиоз
Врожденный ихтиозиформный 0027
Везикулобуллезные и эрозивные заболевания у детей:
Буллезный эпидермолиз
Стафилококковый ошпаренный кожный покров синдром
Буллезное импетиго
Простой герпес
Врожденный эрозивно-везикулярный дерматоз
Аутоиммунные пузырчатые болезни
Генодерматозы:
4 Синдром Шегрена-Ларссона 11
Болезнь накопления нейтральных липидов
Трихотиодистрофия
Синдром Нетертона
Дефицит стероидной сульфатазы
Синдром шелушения кожи
Синдром Конради-Хунерманна-Гаппла
ДЕТСКИЙ синдром
ДЕТСКИЙ синдром
Прогноз
Тяжесть эпидермолитического гиперкератоза варьирует.
Новорожденные с этим заболеванием подвергаются повышенному риску сепсиса, обезвоживания и, в конечном итоге, смерти. Из-за редкости этого заболевания статистические данные о смертности отсутствуют. У тех, кто пережил неонатальный период, эпизоды образования волдырей и кожных инфекций периодически возникают на протяжении всей жизни. В результате у пациентов часто наблюдается значительное обезображивание и резкий запах тела, что вызывает серьезные психологические и социальные расстройства.[1][12]Осложнения
Новорожденные, рожденные с эпидермолитическим гиперкератозом, подвержены более высокому риску обезвоживания, дисбаланса электролитов и сепсиса из-за повышенной трансэпидермальной потери воды и неэффективной барьерной функции кожи. Без надлежащего лечения эти осложнения приводят к летальному исходу. Кроме того, серьезное ладонно-подошвенное поражение может привести к контрактурам пальцев, что негативно влияет на подвижность суставов, походку и осанку.[12]
Сдерживание и обучение пациентов
У пациентов с эпидермолитическим гиперкератозом на протяжении всей жизни наблюдается повышенная хрупкость кожи, поэтому важно обучать пациентов способам сведения к минимуму механической травмы.
Это возможно, если носить свободную, удобную одежду и хорошо сидящую обувь.Улучшение результатов медицинского персонала
Эпидермолитический гиперкератоз обычно проявляется вскоре после рождения. Это неизлечимо, и расстройство прогрессирует. Лечение в первую очередь симптоматическое и зависит от возраста пациента и клинической картины. Врачебное ведение включает семейного врача, педиатра и дерматолога, а также НП и ПА. Младенцы должны находиться под наблюдением в условиях отделения интенсивной терапии для лечения обезвоживания, электролитного дисбаланса и кожной суперинфекции. Сепсис следует лечить внутривенными антибиотиками широкого спектра действия, которые должны быть согласованы с фармацевтом в отношении охвата, дозирования и мониторинга лекарственных взаимодействий. Медсестра должна обучить опекуна или родителя использованию местных смягчающих средств и защитных прокладок для защиты кожи и заживления оголенных участков, которые также могут быть использованы в аптеке.
Поскольку кожа хрупкая, практикующая медсестра должна также информировать пациентов о том, как свести к минимуму механическую травму, что достижимо при ношении свободной, удобной одежды и удобной обуви.Из-за значительной психосоциальной заболеваемости, вызванной отдаленными последствиями эпидермолитического гиперкератоза, необходимо координировать лечение с помощью межпрофессиональной бригады, включающей реаниматологов, инфекционистов, психиатрических медсестер и психиатров. Задержки в диагностике и лечении могут привести к негативным последствиям и возможной смерти. Все эти члены межпрофессиональной медицинской бригады должны сотрудничать и общаться, чтобы добиться положительных результатов лечения пациентов.[1] [Уровень 5]
Контрольные вопросы
Доступ к бесплатным вопросам с несколькими вариантами ответов по этой теме.
Комментарий к этой статье.
Рисунок
Эпидермолитический гиперкератоз. Изображение предоставлено S Bhimji MD
Рисунок
Эпидермолитический гиперкератоз.
Предоставлено д-ром Shyam Verma, MBBS, DVD, FRCP, FAAD, Vadodara, IndiaСсылки
- 1.
Peter Rout D, Nair A, Gupta A, Kumar P. Эпидермолитический гиперкератоз: клиническое обновление. Clin Cosmet Investig Dermatol. 2019;12:333-344. [Статья бесплатно PMC: PMC6512611] [PubMed: 31190940]
- 2.
Covaciu C, Castori M, De Luca N, Ghirri P, Nannipieri A, Ragone G, Zambruno G, Castiglia D. Летальный аутосомно-рецессивный эпидермолитический эпидермолитический из-за новой мутации донорского сайта сплайсинга в KRT10. Бр Дж Дерматол. 2010 июнь; 162 (6): 1384-7. [PubMed: 20302579]
- 3.
Оджи В., Тадини Г., Акияма М., Бланше Бардон С., Бодемер С., Бурра Э., Кудьер П., ДиДжиованна Дж.Дж., Элиас П., Фишер Дж., Флекман П., Джина М., Харпер Дж., Хашимото Т., Хауссер И., Хеннис Х.К., Холь Д., Овнанян А., Исида-Ямамото А., Яцик В.К., Личман С., Ли И., Мазереу-Хотье Дж., Милстон Л., Морис-Пикард Ф., Паллер А.С., Ричард Г.
, Schmuth M, Shimizu H, Sprecher E, Van Steensel M, Taïeb A, Toro JR, Vabres P, Vahlquist A, Williams M, Traupe H. Пересмотренная номенклатура и классификация наследственных ихтиозов: результаты Первой консенсусной конференции по ихтиозу в Сорезе, 2009 г.. J Am Acad Дерматол. 2010 Октябрь; 63 (4): 607-41. [PubMed: 20643494]- 4.
Чипев С.С., Корге Б.П., Маркова Н., Бейл С.Дж., ДиДжиованна Д.Дж., Комптон Д.Г., Штайнерт П.М. Мутация лейцина—-пролина в субдомене h2 кератина 1 вызывает эпидермолитический гиперкератоз. Клетка. 1992 Сентябрь 04; 70 (5): 821-8. [PubMed: 1381288]
- 5.
Исида-Ямамото А., МакГрат Дж.А., судья М.Р., Ли И.М., Лейн Э.Б., Иди Р.А. Избирательное участие кератинов К1 и К10 в цитоскелетной аномалии эпидермолитического гиперкератоза (буллезная врожденная ихтиозиформная эритродермия). Джей Инвест Дерматол. 1992 июля; 99(1):19-26. [PubMed: 1376754]
- 6.
Виртанен М., Смит С.К., Гедде-Даль Т.
, Валквист А., Боуден П.Е. Мутации сайта сплайсинга и делеции в генах кератина (KRT1 и KRT10): необычные фенотипические изменения у скандинавских пациентов с эпидермолитическим гиперкератозом. Джей Инвест Дерматол. 2003 ноябрь; 121(5):1013-20. [PubMed: 14708600]- 7.
Ирвин А.Д., Маклин В.Х. Кератиновые заболевания человека: увеличивающийся спектр заболеваний и тонкость корреляции фенотип-генотип. Бр Дж Дерматол. 1999 мая; 140(5):815-28. [PubMed: 10354017]
- 8.
Мюллер Ф.Б., Хубер М., Кинаджян Т., Хауссер И., Шаффрат С., Криг Т., Холь Д., Корге Б.П., Арин М.Дж. Нокаут кератина 10 человека вызывает рецессивный эпидермолитический гиперкератоз. Хум Мол Жене. 01 апреля 2006 г.; 15 (7): 1133-41. [PubMed: 16505000]
- 9.
Цубота А., Акияма М., Канитакис Дж., Сакаи К., Номура Т., Клауди А., Симидзу Х. Легкая рецессивная буллезная врожденная ихтиозиформная эритродермия из-за ранее невыявленной гомозиготной мутации 10 .
Джей Инвест Дерматол. 2008 июль; 128 (7): 1648-52. [В паблике: 18219278]- 10.
Ди Джованна Дж.Дж., Бэйл С.Дж. Клиническая гетерогенность эпидермолитического гиперкератоза. Арка Дерматол. 1994 г., август; 130(8):1026-35. [PubMed: 8053700]
- 11.
Paller AS, Syder AJ, Chan YM, Yu QC, Hutton E, Tadini G, Fuchs E. Генетический и клинический мозаицизм в типе эпидермального невуса. N Engl J Med. 1994 24 ноября; 331 (21): 1408-15. [PubMed: 7526210]
- 12.
Квак Дж., Маверакис Э. Эпидермолитический гиперкератоз. Dermatol Online J. 2006, 08 сентября; 12 (5): 6. [В паблике: 16962021]
- 13.
Fuchs E, Coulombe P, Cheng J, Chan YM, Hutton E, Syder A, Degenstein L, Yu QC, Letai A, Vassar R. Генетические основы простого буллезного эпидермолиза и эпидермолитического гиперкератоза. Джей Инвест Дерматол. 1994 г., ноябрь; 103 (5 доп.): 25S-30S. [PubMed: 7525738]
- 14.
Nelson WG, Sun TT.
Классы кератина 50 и 58 кДа как молекулярные маркеры многослойного плоского эпителия: исследования клеточных культур. Джей Селл Биол. 1983 июль; 97(1):244-51. [Бесплатная статья PMC: PMC2112495] [PubMed: 6190820]- 15.
Roop DR, Huitfeldt H, Kilkenny A, Yuspa SH. Регулируемая экспрессия кератинов, связанных с дифференцировкой, в культивируемых клетках эпидермиса, обнаруженная моноспецифическими антителами к уникальным пептидам эпидермальных кератинов мыши. Дифференциация. 1987;35(2):143-50. [PubMed: 2450799]
- 16.
Syder AJ, Yu QC, Paller AS, Giudice G, Pearson R, Fuchs E. Генетические мутации в генах K1 и K10 у пациентов с эпидермолитическим гиперкератозом. Взаимосвязь между локализацией и тяжестью заболевания. Джей Клин Инвест. 1994 апреля; 93 (4): 1533-42. [Бесплатная статья PMC: PMC294170] [PubMed: 7512983]
- 17.
Chen PJ, Li CX, Wen J, Peng YS, Zeng K, Zhang SQ, Tian X, Zhang XB. Мутация S159P гена кератина 10 вызывает тяжелую форму эпидермолитического гиперкератоза.
 Новорожденные с этим заболеванием подвергаются повышенному риску сепсиса, обезвоживания и, в конечном итоге, смерти. Из-за редкости этого заболевания статистические данные о смертности отсутствуют. У тех, кто пережил неонатальный период, эпизоды образования волдырей и кожных инфекций периодически возникают на протяжении всей жизни. В результате у пациентов часто наблюдается значительное обезображивание и резкий запах тела, что вызывает серьезные психологические и социальные расстройства.[1][12]
Новорожденные с этим заболеванием подвергаются повышенному риску сепсиса, обезвоживания и, в конечном итоге, смерти. Из-за редкости этого заболевания статистические данные о смертности отсутствуют. У тех, кто пережил неонатальный период, эпизоды образования волдырей и кожных инфекций периодически возникают на протяжении всей жизни. В результате у пациентов часто наблюдается значительное обезображивание и резкий запах тела, что вызывает серьезные психологические и социальные расстройства.[1][12] Это возможно, если носить свободную, удобную одежду и хорошо сидящую обувь.
Это возможно, если носить свободную, удобную одежду и хорошо сидящую обувь.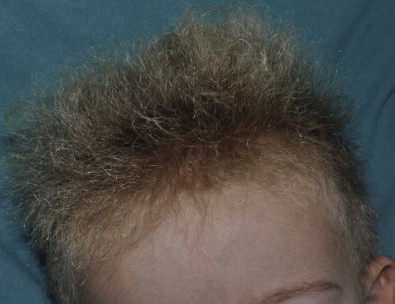 Поскольку кожа хрупкая, практикующая медсестра должна также информировать пациентов о том, как свести к минимуму механическую травму, что достижимо при ношении свободной, удобной одежды и удобной обуви.
Поскольку кожа хрупкая, практикующая медсестра должна также информировать пациентов о том, как свести к минимуму механическую травму, что достижимо при ношении свободной, удобной одежды и удобной обуви. Предоставлено д-ром Shyam Verma, MBBS, DVD, FRCP, FAAD, Vadodara, India
Предоставлено д-ром Shyam Verma, MBBS, DVD, FRCP, FAAD, Vadodara, India , Schmuth M, Shimizu H, Sprecher E, Van Steensel M, Taïeb A, Toro JR, Vabres P, Vahlquist A, Williams M, Traupe H. Пересмотренная номенклатура и классификация наследственных ихтиозов: результаты Первой консенсусной конференции по ихтиозу в Сорезе, 2009 г.. J Am Acad Дерматол. 2010 Октябрь; 63 (4): 607-41. [PubMed: 20643494]
, Schmuth M, Shimizu H, Sprecher E, Van Steensel M, Taïeb A, Toro JR, Vabres P, Vahlquist A, Williams M, Traupe H. Пересмотренная номенклатура и классификация наследственных ихтиозов: результаты Первой консенсусной конференции по ихтиозу в Сорезе, 2009 г.. J Am Acad Дерматол. 2010 Октябрь; 63 (4): 607-41. [PubMed: 20643494] , Валквист А., Боуден П.Е. Мутации сайта сплайсинга и делеции в генах кератина (KRT1 и KRT10): необычные фенотипические изменения у скандинавских пациентов с эпидермолитическим гиперкератозом. Джей Инвест Дерматол. 2003 ноябрь; 121(5):1013-20. [PubMed: 14708600]
, Валквист А., Боуден П.Е. Мутации сайта сплайсинга и делеции в генах кератина (KRT1 и KRT10): необычные фенотипические изменения у скандинавских пациентов с эпидермолитическим гиперкератозом. Джей Инвест Дерматол. 2003 ноябрь; 121(5):1013-20. [PubMed: 14708600] Джей Инвест Дерматол. 2008 июль; 128 (7): 1648-52. [В паблике: 18219278]
Джей Инвест Дерматол. 2008 июль; 128 (7): 1648-52. [В паблике: 18219278]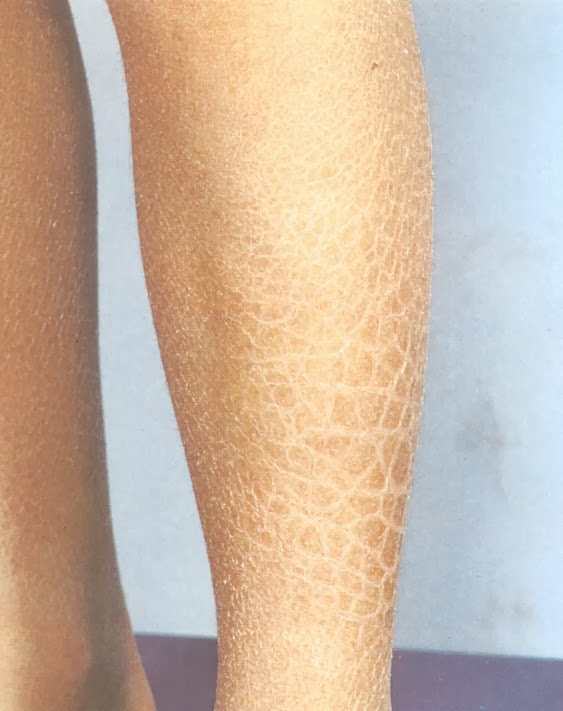 Классы кератина 50 и 58 кДа как молекулярные маркеры многослойного плоского эпителия: исследования клеточных культур. Джей Селл Биол. 1983 июль; 97(1):244-51. [Бесплатная статья PMC: PMC2112495] [PubMed: 6190820]
Классы кератина 50 и 58 кДа как молекулярные маркеры многослойного плоского эпителия: исследования клеточных культур. Джей Селл Биол. 1983 июль; 97(1):244-51. [Бесплатная статья PMC: PMC2112495] [PubMed: 6190820] J Eur Acad Dermatol Venereol. 2016 Окт;30(10):e102-e104. [PubMed: 26373619]
J Eur Acad Dermatol Venereol. 2016 Окт;30(10):e102-e104. [PubMed: 26373619]
 [1][2][3]
[1][2][3]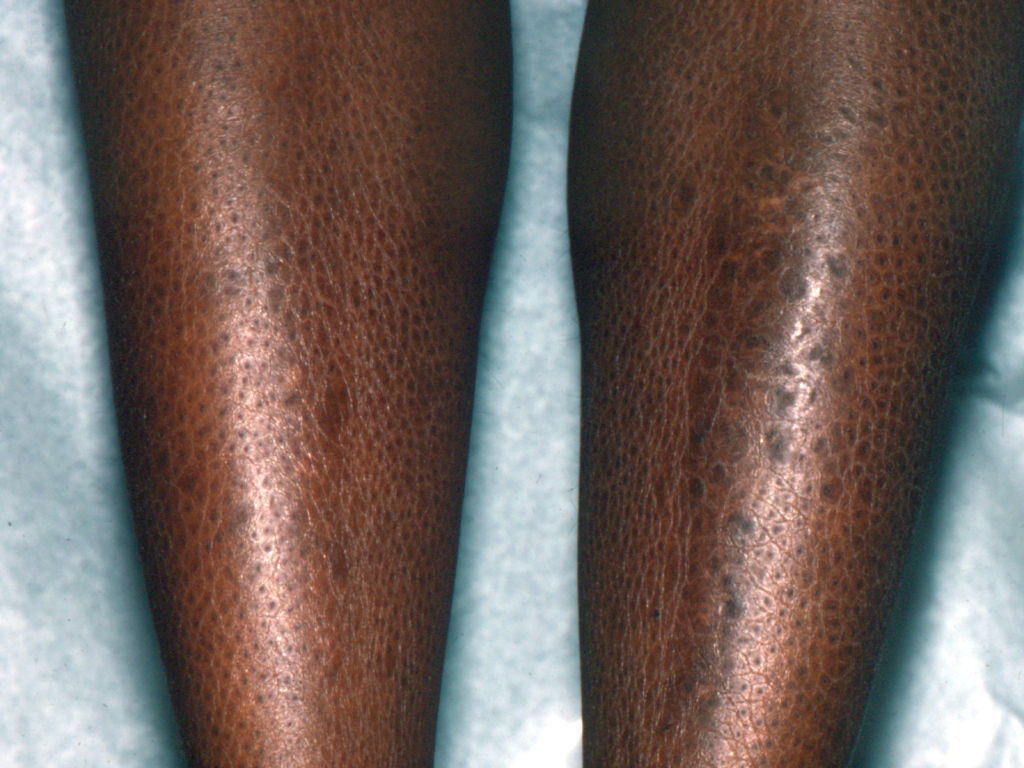 Если эти мутации затрагивают клетки гонад, они могут передаваться потомству и вызывать фенотипический эпидермолитический гиперкератоз.[7][11] В настоящее время эпидермолитический гиперкератоз является единственным известным кератиновым заболеванием человека, проявляющим генетический мозаицизм.[7]
Если эти мутации затрагивают клетки гонад, они могут передаваться потомству и вызывать фенотипический эпидермолитический гиперкератоз.[7][11] В настоящее время эпидермолитический гиперкератоз является единственным известным кератиновым заболеванием человека, проявляющим генетический мозаицизм.[7] Более толстые пучки тонофиламентов обеспечивают повышенную выживаемость кератинов.[1][14][15]
Более толстые пучки тонофиламентов обеспечивают повышенную выживаемость кератинов.[1][14][15]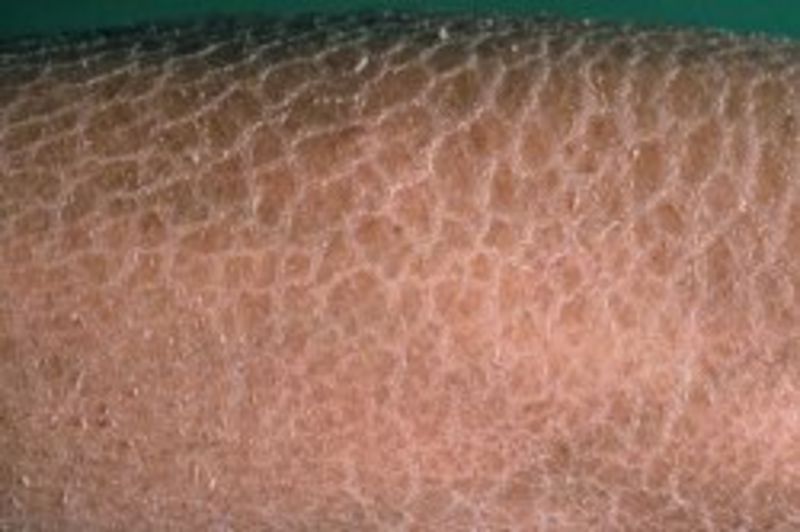 Снайдер и др. идентифицировали мутацию аргинина в гистидин на амино-конце а-спирального стержневого домена KRT10 и мутацию тирозина в цистеин в карбокси-домене KRT1. Тяжелые формы эпидермолитического гиперкератоза связаны с делецией экзона 6 гена KRT1[6] и миссенс-мутациями (c.475T>C;p.Ser159).Pro и c.562A>C;p.N188H) в KRT10 и KRT1 соответственно.[1][17]
Снайдер и др. идентифицировали мутацию аргинина в гистидин на амино-конце а-спирального стержневого домена KRT10 и мутацию тирозина в цистеин в карбокси-домене KRT1. Тяжелые формы эпидермолитического гиперкератоза связаны с делецией экзона 6 гена KRT1[6] и миссенс-мутациями (c.475T>C;p.Ser159).Pro и c.562A>C;p.N188H) в KRT10 и KRT1 соответственно.[1][17] [12] У пациентов с мозаичной формой эпидермолитического гиперкератоза обнаруживаются фокальные очаги поражения с пропуском участков нормального эпидермиса.[1][2]
[12] У пациентов с мозаичной формой эпидермолитического гиперкератоза обнаруживаются фокальные очаги поражения с пропуском участков нормального эпидермиса.[1][2] Обычно сопутствующие симптомы включают ксероз, зуд, болезненные трещины, ангидроз и уменьшение объема движений в суставах.[1][12]
Обычно сопутствующие симптомы включают ксероз, зуд, болезненные трещины, ангидроз и уменьшение объема движений в суставах.[1][12]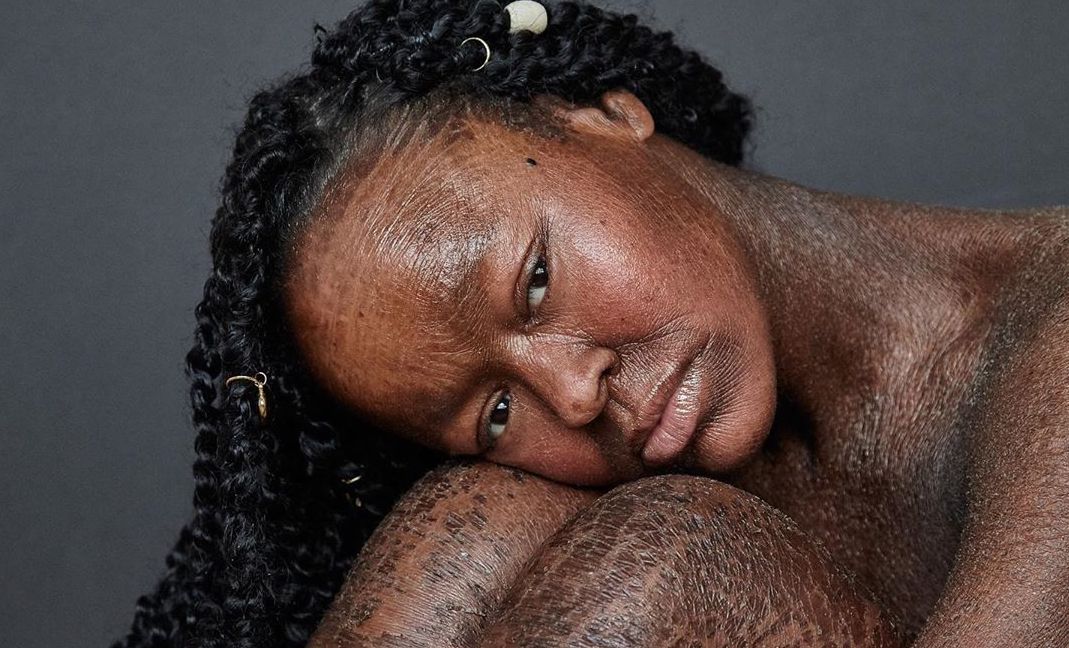 Анализ генетических мутаций дефектов кератина с помощью мультигенного скрининга в настоящее время является золотым стандартом и может использоваться для пренатального скрининга.[1][12] Пренатальную диагностику можно получить с помощью забора ворсин хориона, амниоцентеза и фетоскопии с биопсией кожи плода. Однако эти исследования имеют ограничения низкой чувствительности или генетической гетерогенности. Оптические и спектроскопические методы, такие как рамановская спектроскопия и оптическая когерентная томография, использовались для идентификации немеланомного рака кожи in vivo и могут быть потенциальным методом диагностики эпидермолитического гиперкератоза.
Анализ генетических мутаций дефектов кератина с помощью мультигенного скрининга в настоящее время является золотым стандартом и может использоваться для пренатального скрининга.[1][12] Пренатальную диагностику можно получить с помощью забора ворсин хориона, амниоцентеза и фетоскопии с биопсией кожи плода. Однако эти исследования имеют ограничения низкой чувствительности или генетической гетерогенности. Оптические и спектроскопические методы, такие как рамановская спектроскопия и оптическая когерентная томография, использовались для идентификации немеланомного рака кожи in vivo и могут быть потенциальным методом диагностики эпидермолитического гиперкератоза. Для защиты кожи и заживления оголенных участков следует использовать местные смягчающие средства и защитные прокладки.[12]
Для защиты кожи и заживления оголенных участков следует использовать местные смягчающие средства и защитные прокладки.[12] Таким образом, рекомендуется использовать низкие начальные дозы, а затем постепенно увеличивать их под тщательным контролем [19].] Следует отметить, что пациенты с мутациями кератина 10 лучше реагируют на местные или системные ретиноиды по сравнению с пациентами с мутациями кератина 1.[1][12] Антибактериальные мыла, хлоргексидин и ванны с разбавленным гипохлоритом натрия могут уменьшить бактериальную колонизацию. При возникновении бактериальной инфекции кожи необходимы местные или системные антибиотики.[1][12]
Таким образом, рекомендуется использовать низкие начальные дозы, а затем постепенно увеличивать их под тщательным контролем [19].] Следует отметить, что пациенты с мутациями кератина 10 лучше реагируют на местные или системные ретиноиды по сравнению с пациентами с мутациями кератина 1.[1][12] Антибактериальные мыла, хлоргексидин и ванны с разбавленным гипохлоритом натрия могут уменьшить бактериальную колонизацию. При возникновении бактериальной инфекции кожи необходимы местные или системные антибиотики.[1][12] Новорожденные с этим заболеванием подвергаются повышенному риску сепсиса, обезвоживания и, в конечном итоге, смерти. Из-за редкости этого заболевания статистические данные о смертности отсутствуют. У тех, кто пережил неонатальный период, эпизоды образования волдырей и кожных инфекций периодически возникают на протяжении всей жизни. В результате у пациентов часто наблюдается значительное обезображивание и резкий запах тела, что вызывает серьезные психологические и социальные расстройства.[1][12]
Новорожденные с этим заболеванием подвергаются повышенному риску сепсиса, обезвоживания и, в конечном итоге, смерти. Из-за редкости этого заболевания статистические данные о смертности отсутствуют. У тех, кто пережил неонатальный период, эпизоды образования волдырей и кожных инфекций периодически возникают на протяжении всей жизни. В результате у пациентов часто наблюдается значительное обезображивание и резкий запах тела, что вызывает серьезные психологические и социальные расстройства.[1][12]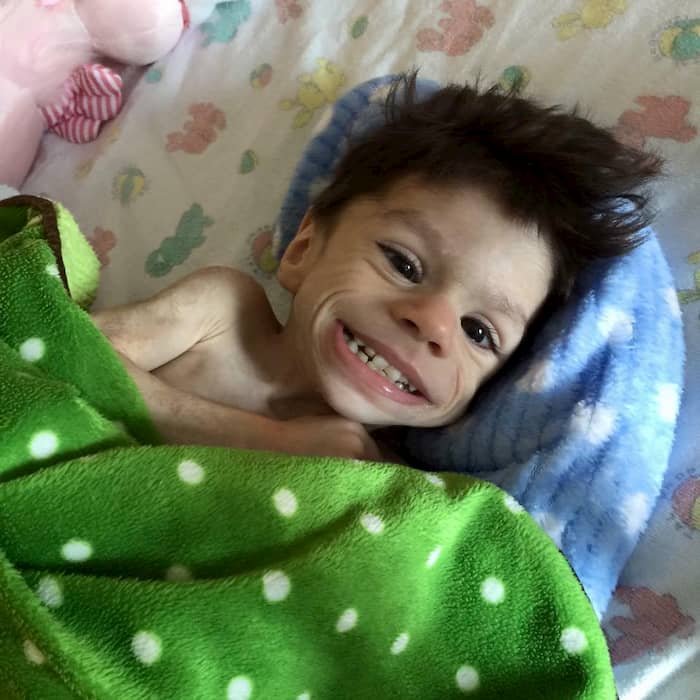 Это возможно, если носить свободную, удобную одежду и хорошо сидящую обувь.
Это возможно, если носить свободную, удобную одежду и хорошо сидящую обувь. Поскольку кожа хрупкая, практикующая медсестра должна также информировать пациентов о том, как свести к минимуму механическую травму, что достижимо при ношении свободной, удобной одежды и удобной обуви.
Поскольку кожа хрупкая, практикующая медсестра должна также информировать пациентов о том, как свести к минимуму механическую травму, что достижимо при ношении свободной, удобной одежды и удобной обуви. Предоставлено д-ром Shyam Verma, MBBS, DVD, FRCP, FAAD, Vadodara, India
Предоставлено д-ром Shyam Verma, MBBS, DVD, FRCP, FAAD, Vadodara, India/img2.papilot.pl/800w/2014/10/rybia-luska-arlekinowa-86343067.jpg) , Schmuth M, Shimizu H, Sprecher E, Van Steensel M, Taïeb A, Toro JR, Vabres P, Vahlquist A, Williams M, Traupe H. Пересмотренная номенклатура и классификация наследственных ихтиозов: результаты Первой консенсусной конференции по ихтиозу в Сорезе, 2009 г.. J Am Acad Дерматол. 2010 Октябрь; 63 (4): 607-41. [PubMed: 20643494]
, Schmuth M, Shimizu H, Sprecher E, Van Steensel M, Taïeb A, Toro JR, Vabres P, Vahlquist A, Williams M, Traupe H. Пересмотренная номенклатура и классификация наследственных ихтиозов: результаты Первой консенсусной конференции по ихтиозу в Сорезе, 2009 г.. J Am Acad Дерматол. 2010 Октябрь; 63 (4): 607-41. [PubMed: 20643494] , Валквист А., Боуден П.Е. Мутации сайта сплайсинга и делеции в генах кератина (KRT1 и KRT10): необычные фенотипические изменения у скандинавских пациентов с эпидермолитическим гиперкератозом. Джей Инвест Дерматол. 2003 ноябрь; 121(5):1013-20. [PubMed: 14708600]
, Валквист А., Боуден П.Е. Мутации сайта сплайсинга и делеции в генах кератина (KRT1 и KRT10): необычные фенотипические изменения у скандинавских пациентов с эпидермолитическим гиперкератозом. Джей Инвест Дерматол. 2003 ноябрь; 121(5):1013-20. [PubMed: 14708600] Джей Инвест Дерматол. 2008 июль; 128 (7): 1648-52. [В паблике: 18219278]
Джей Инвест Дерматол. 2008 июль; 128 (7): 1648-52. [В паблике: 18219278] Классы кератина 50 и 58 кДа как молекулярные маркеры многослойного плоского эпителия: исследования клеточных культур. Джей Селл Биол. 1983 июль; 97(1):244-51. [Бесплатная статья PMC: PMC2112495] [PubMed: 6190820]
Классы кератина 50 и 58 кДа как молекулярные маркеры многослойного плоского эпителия: исследования клеточных культур. Джей Селл Биол. 1983 июль; 97(1):244-51. [Бесплатная статья PMC: PMC2112495] [PubMed: 6190820]
