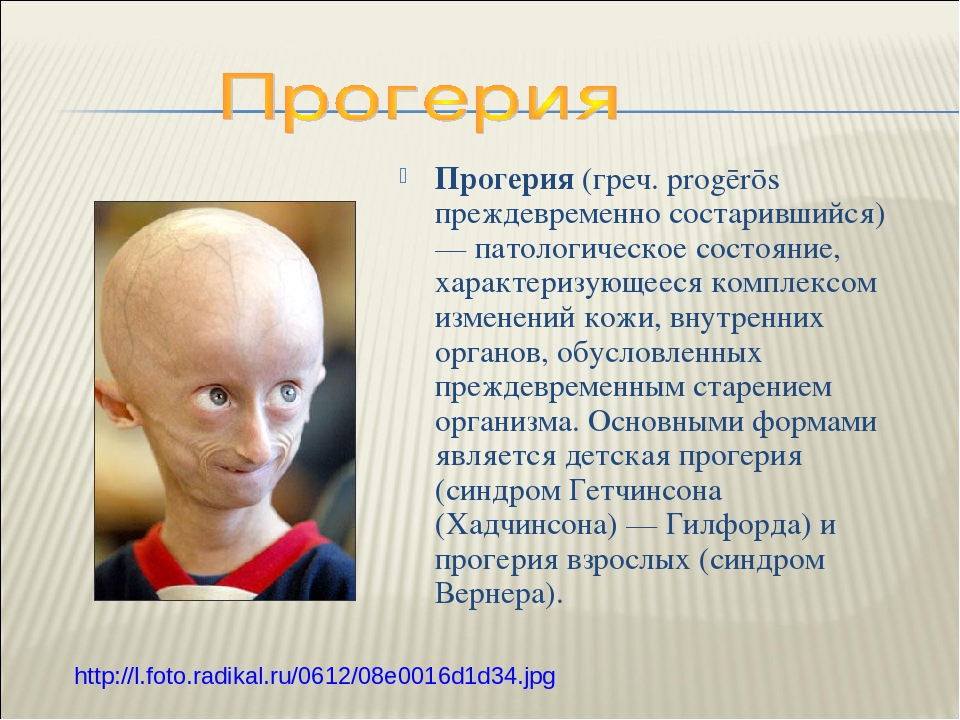
Прогерия (Синдром Вернера) — санатории где лечат, цены и отзывы
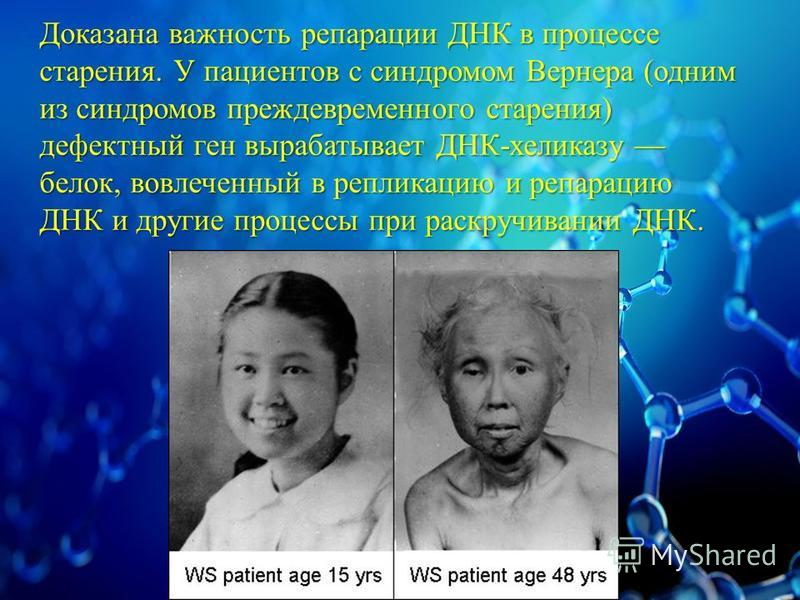
Прогерия (Синдром Вернера) — это генетическое заболевание, характеризующееся преждевременным старением больного. У него отмечаются поражения различных систем организма, появление атеросклероза и злокачественных опухолей. Человек очень быстро превращается в старика. Чаще прогерией болеют мужчины.
Причины
Синдром Вернера передается по наследству, это аутосомно-рецессивный тип заболевания. В данном случае ребенок рождается от родителей, имеющих дефектный ген WRN, что приводит к нарушению обмена соединительной ткани. Если подобная мутация есть только у одного родителя, болезнь не возникает, если же у обоих, то у ребенка развивается прогерия.
Симптомы
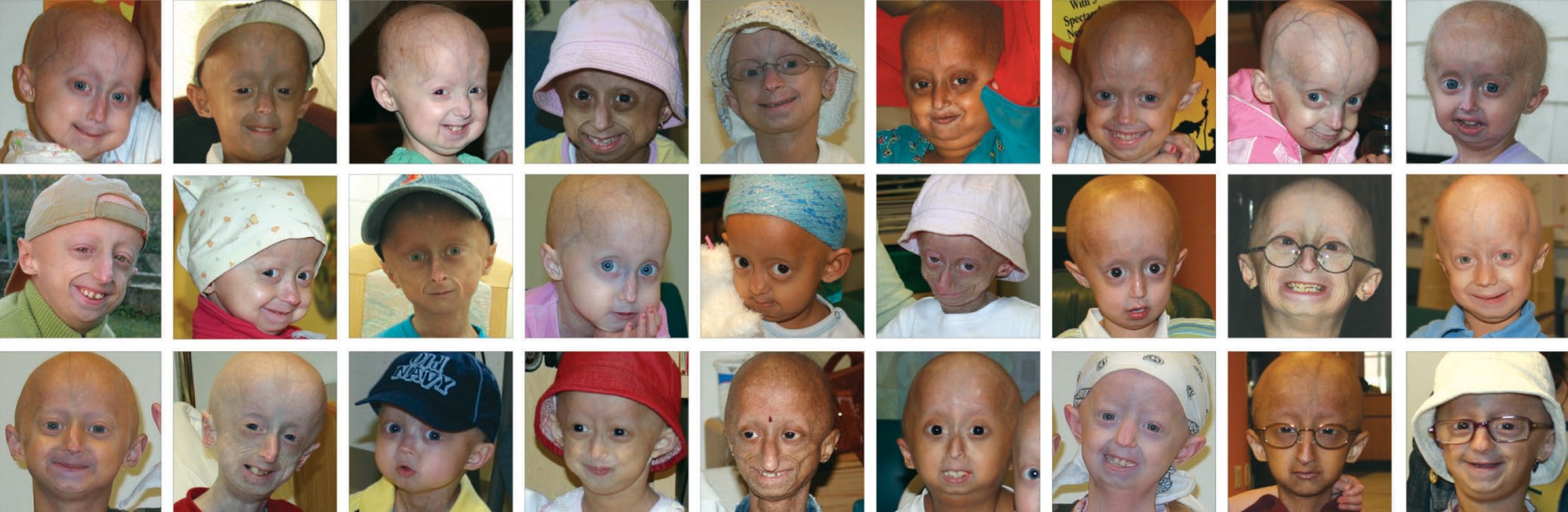
Признаки заболевания обычно появляются в 14-18 лет. В этом возрасте наблюдается седина, выпадение волос. Кожа становится бледной и плотной, с пигментными пятнами, появляются морщины. Мышцы и подкожная жировая клетчатка атрофируются. Руки и ноги становятся непропорционально тонкими.
На третьем десятилетии после начала болезни у пациента меняется голос, он становится высоким. Возникают следующие симптомы:
- Катаракта.
- Сухость кожи.
- Мозоли на подошвах.
- Язвы на нижних конечностях.
- Низкий рост.
- Лунообразное лицо с выступающим подбородком и узким ртом.
- Остеопороз.
- Гиперпигментация.
- Деформация кистей.
- Остеоартриты.
- Плоскостопия, стерильность и так далее.
В зрелом возрасте больные выглядят как очень пожилые люди. На четвертом десятилетии после возникновения прогерии у них диагностируется сахарный диабет, атеросклероз, дисфункции щитовидной железы. В ряде случаев возможно развитие раковых заболеваний.
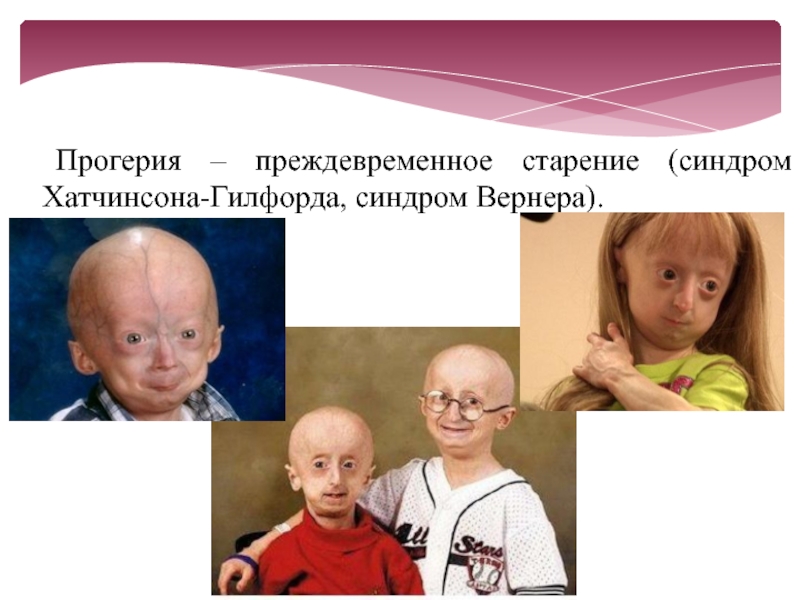
Виды/формы
Синдром Вернера является подвидом прогерии, он возникает у людей в период полового созревания и даже позже. Другой подвид прогерии — синдром Хатчинсона-Гилфорда. Эта болезнь возникает у маленьких детей.
Стадии
Болезнь развивается постепенно, в течение нескольких десятилетий. На начальной стадии наблюдается признаки раннего старения: седина в волосах и их впадание. С течением времени больной теряет способность вести полноценную жизнь. Заканчивается прогерия смертью больного. Обычно это наступает в возрасте 50 лет.
Методы лечения
Данная болезнь является хронической и постоянно прогрессирует, поэтому лечение сводится к профилактике осложнений прогерии. Больные регулярно наблюдаются у эндокринологов, кардиологов и так далее. Это поможет замедлить развитие патологических изменений в организме. Излечиться от синдрома Вернера невозможно, пациент обычно умирает от раковых опухолей или последствий атеросклероза.
Профилактика в санаториях России
Больным с синдромом Вернера будет полезно санаторно-курортное лечение. В России есть немало санаториев с комфортными условиями проживания, прекрасным оснащением, где пациенты смогут приостановить проявления болезни и укрепят свой организм. В этом им помогут опытные врачи и новые методики лечения.
В этом им помогут опытные врачи и новые методики лечения.
Синдромы преждевременного старения | Системная схема старения человека
СИНДРОМЫ ПРЕЖДЕВРЕМЕННОГО СТАРЕНИЯ
Пожалуй наиболее ярким доказательством определяющей роли генов в старении являются моногенные болезни с признаками ускоренного старения (прогерии). О причинах данных заболеваний и их связи с естественным старением и пойдет речь.
Прогерии
Одним из подходов к изучению молекулярных основ старения человека является выяснение причин заболеваний преждевременного старения — так называемых частичных прогерий. В большинстве своем они моногенны, а значит, легко поддаются анализу. Недостатком данного подхода является то, что иногда их симптомы лишь напоминают свойства нормального старения, либо представлены не все свойства. Например, симптомы старения при прогериях более выражены и могут появляться в другой последовательности, чем в случае с «нормальным» старением. В частности, рост ногтей замедляется при старении, тогда как при прогериях с короткими теломерами останавливается полностью.
Таким образом, мутации определенных генов у человека приводят к тяжелым заболеваниям, связанным с признаками преждевременного старения. Что это за заболевания и какие гены их обусловливают? Найдем ответы на эти вопросы.
Синдром Вернера
Одним из наиболее известных заболеваний с признаками ускоренного старения является синдром Вернера (http://en.wikipedia.org/wiki/Werner_syndrome, прогерия взрослых) — аутосомно-рецессивное заболевание (то есть контролируется рецессивными аллелями аутосомного гена), характеризующееся проявлением симптомов преждевременного старения кожи, сосудистой и репродуктивной системы, костей. До полового созревания пациенты развиваются нормально. Симптомы старения у них начинаются в ранней зрелости. Уже в молодом возрасте они страдают от катаракт, склеродермальных и дегенеративных сосудистых изменений, диабета и атеросклероза, остеопороза, высокой частоты некоторых видов рака, поседения.
Функция гена данного заболевания была охарактеризована первой среди генов всех прогерий (пионерская статья). При синдроме Вернера аутосомно-рецессивная мутация в гене WRN, находящемся на хромосоме 8, приводит к нарушению функции особой ДНК-геликазы. Основная роль белка WRN в клетке — реинициирование блокированных репликационных вилок. В результате мутации вызывается нарушение репликации и репарации ДНК, экспрессии генов, ускоренное укорочение теломер и повышенная чувствительность клеток к апоптозу (обзор).
ДНК-геликазы
Здесь стоит напомнить, что такое ДНК-геликазы. Данные ферменты расплетают двухцепочечную ДНК, что является необходимым условием для большинства молекулярно-генетических процессов, таких как синтез копий ДНК (репликация), а также транскрипция РНК и репарация ДНК.
Синдром Ротмунда-Томпсона (RTS)
Для генетически близкого заболевания, аутосомно-рецессивного синдрома Ротмунда-Томпсона (http://en. wikipedia.org/wiki/Rothmund-Thompson’s_syndrome), характерно наличие особой гиперпигментации кожи (пойкилодермия), гиперчувствительности к солнечным лучам, задержки роста, гипогонадотропного гипогонадизма, анемии, контрактуры мягких тканей, гиподонтии, ювенильных катаракт, проблем с ростом волос, остеогенной саркомы (последнее заболевание является отличительным признаком и при синдроме Вернера). Как и ген предыдущего заболевания, ген данного заболевания (RECQL4) относится к семейству RecQ 3′-5′ ДНК геликаз, которые участвуют в поддержании стабильности генома через регуляцию репликационной вилки.
wikipedia.org/wiki/Rothmund-Thompson’s_syndrome), характерно наличие особой гиперпигментации кожи (пойкилодермия), гиперчувствительности к солнечным лучам, задержки роста, гипогонадотропного гипогонадизма, анемии, контрактуры мягких тканей, гиподонтии, ювенильных катаракт, проблем с ростом волос, остеогенной саркомы (последнее заболевание является отличительным признаком и при синдроме Вернера). Как и ген предыдущего заболевания, ген данного заболевания (RECQL4) относится к семейству RecQ 3′-5′ ДНК геликаз, которые участвуют в поддержании стабильности генома через регуляцию репликационной вилки.
Синдром Блума
При аутосомно-рецессивном синдроме Блума (Bloom syndrome) отмечены гиперчувствительность к ультрафиолету, иммунодефицит, малорослость, остеосаркомы (являющиеся причиной смерти до 30 лет у больных с данным синдромом). Характерные для старения признаки менее выражены, чем при предыдущих синдромах, например, наблюдается преждевременная менопауза у женщин. Вследствие мутации в гене BLM, принадлежащем к генам ДНК-геликаз, синдром характеризуется нестабильностью генома и повышенным риском канцерогенеза.
Синдром Хатчинсона-Гилфорда
Очень часто, когда говорят о прогериях, имеют в виду именно синдром Хатчинсона-Гилфорда (Hutchinson-Gilford syndrome), так называемую «прогерию детей». Это крайне редкое заболевание (<1/1000000, тогда как частота предыдущих прогерий составляет в среднем < 1/100000). Еще одним отличием данной прогерии является то, что мутация, вызывающая ее, всегда возникает de novo, то есть не наследуется. Это не удивительно, поскольку носители погибают до репродуктивного возраста. Дети шестилетнего возраста при синдроме Хатчинсона-Гилфорда выглядят как уже пожилые люди и погибают от сильного атеросклероза к 13 годам. Данное заболевание отличают неспособность к росту, липоатрофия, костные нарушения, маленький клювообразный нос, срезанный подбородок, полная потеря волос, пятнистая гипопигментация кожи. С развитием заболевания возникают атеросклеротические бляшки, которые становятся проникающими, приводя к сердечным приступам и смерти.
 Мутация приводит к синтезу укороченной версии белка и, соответственно, к недостатку количества ламина А дикого типа.
Мутация приводит к синтезу укороченной версии белка и, соответственно, к недостатку количества ламина А дикого типа.Прогерия Хатчинсона-Джилфорда сопровождается дефектами в ядерной структуре и функциях: присутствует дизморфия поверхности ядра, увеличение уровня повреждений ДНК, снижение экспрессии ряда ядерных белков, включая важные гетерохроматиновые белки HP1 и LAP2 (из группы ламин А-ассоциированных белков). Кроме того, в клетках больных нарушен паттерн модифицированных гистонов: происходит снижение гетерохроматин-специфичного триметилирования по остатку Lys9 в гистоне h4 (статья). Таким образом, ядра клеток больных прогерией Хатчинсона-Джилфорда теряют гетерохроматин. В результате происходит патологическая сверхактивация ряда транскриптов, в норме репрессированных, например перицентрического сателлитного повтора III (статья). Коррекция сплайсинга ламина А в клетках пациентов восстанавливает: нормальную морфологию ядра, гетерохроматин-специфичную модификацию гистонов, экспрессию ряда дисрегулированых генов (статья).
Структура ядра
Таким образом, молекулярной причиной данного синдрома являются нарушения структуры ядра. Ядро клетки высших организмов является сложным, высокоорганизованным хранилищем индивидуальной генетической информации. Типичное ядро содержит особые функциональные области, представленные упорядоченно расположенными хромосомами и белковыми субкомпартментами, в которых происходят специфические процессы, включая экспрессию генов. В структурной организации ядра важную роль играет ядерная ламина. Она состоит из белков ламинов А и В типа. Эти промежуточные филаментные белки формируют переплетеную сеть, расположенную на периферии ядра и подстилающую ядерную мембрану. Ламина играет регуляторную роль в экспрессии генов, поскольку белки ламины взаимодействуют с хроматином и могут участвовать в фиксации и организации участков генома в пространстве. Ламина обеспечивает механические и поверхностные свойства ядра и является участком стыковки с периферическим гетерохроматином.
Рестриктивная дермопатия
Рестриктивная дермопатия (Restrictive dermopathy) представляет собой редкий аутосомно-рецессивный синдром. Это летальное перинатальное прогероидное заболевание, характеризуемое задержкой роста, плотной и жесткой кожей, облысением, микрогнатией и другими нарушениями костей, вызывается делецией гена ZMPSTE24, кодирующего протеазу, необходимую для эндопротеолитического процессинга преламина А в зрелый ламин. Таким образом, как и синдром Хатчинсона-Гилфорда, она вызывается дефектом биогенеза ламина А.
Пигментная ксеродерма
Есть целая группа генетических заболеваний, связанных с дефектами различных форм репарации ДНК и имеющих отдельные симптомы, указывающие на процессы ускоренного старения.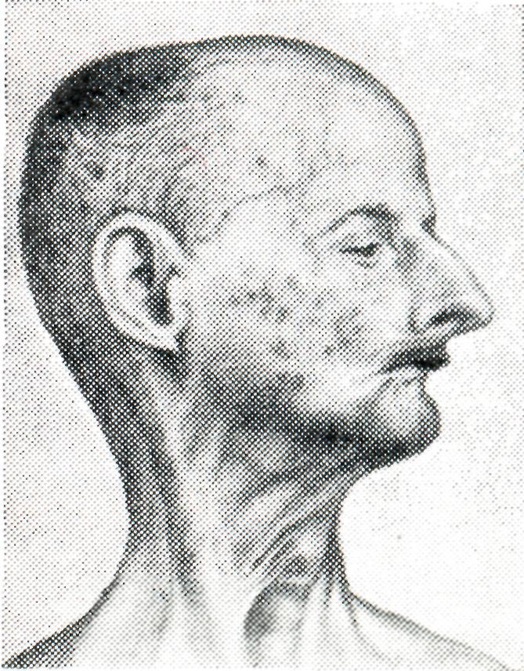 Среди таких патологий одной из наиболее изученных является пигментная ксеродерма (Xeroderma pigmentosum) — редкое аутосомно-рецессивное заболевание, характеризуемое гиперчувствительностью к свету, ненормальной пигментацией и предрасположенностью к раку кожи, особенно на подверженных солнцу участках тела. Имеются различные симптомы от умеренных кожных нарушений до тяжелых повреждений кожи. Данное заболевание вызывается генетическими дефектами раннего этапа эксцизионной репарации нуклеотидов. Существует семь комплементарных групп генов пигментной ксеродермы (от XPA до XPG). Группа G (XPG) кодирует белок с молекулярной массой 133 кДа, который имеет структура-специфичную эндонуклеазную активность и функционирует как 3′-нуклеаза в реакции двойного разрезания при эксцизионной репарации нуклеотидов. Первичные культуры эмбриональных фибробластов, полученные от XPG-дефицитных мышей, характеризуются активацией транскрипционного фактора р53 и преждевременным старением. Гены ксеродермы группы B и D (XPB, XPD) кодируют субъединицы транскрипционного фактора TFIIH.
Среди таких патологий одной из наиболее изученных является пигментная ксеродерма (Xeroderma pigmentosum) — редкое аутосомно-рецессивное заболевание, характеризуемое гиперчувствительностью к свету, ненормальной пигментацией и предрасположенностью к раку кожи, особенно на подверженных солнцу участках тела. Имеются различные симптомы от умеренных кожных нарушений до тяжелых повреждений кожи. Данное заболевание вызывается генетическими дефектами раннего этапа эксцизионной репарации нуклеотидов. Существует семь комплементарных групп генов пигментной ксеродермы (от XPA до XPG). Группа G (XPG) кодирует белок с молекулярной массой 133 кДа, который имеет структура-специфичную эндонуклеазную активность и функционирует как 3′-нуклеаза в реакции двойного разрезания при эксцизионной репарации нуклеотидов. Первичные культуры эмбриональных фибробластов, полученные от XPG-дефицитных мышей, характеризуются активацией транскрипционного фактора р53 и преждевременным старением. Гены ксеродермы группы B и D (XPB, XPD) кодируют субъединицы транскрипционного фактора TFIIH.
Трихотиодистрофия
Заболевание трихотиодистрофия (Trichothiodystrophy) проявляется в пониженном содержании серы в волосах и в их повышенной ломкости, задержке умственного развития, гиперчувствительности к ультрафиолету и в других врожденных аномалиях. Причина заболевания — мутации в генах XPB и XPD, кодирующих субъединицы транскрипционного фактора TFIIH. Продукты генов XPB и XPD имеют геликазную и АТФ-азную активность и участвуют в формировании «транскрипционного пузырька», а также в эксцизионной репарации нуклеотидов, связанной с транскрипцией (Transcription-coupled repair).
Синдром Кокейна
Синдром Кокейна (Cockayne syndrome) — редкое наследственное заболевание, при котором индивид не способен к росту, обладает короткой продолжительностью жизни и нейрологической дисфункцией. Клетки больных синдромом Кокейна чувствительны к ультрафиолету и окислительным формам повреждения ДНК, поскольку характеризуются дефектом одного из механизмов эксцизионной репарации нуклеотидов — связанного с транскрипцией репарационного процесса, предназначенного для удаления крупных повреждений транскрибируемых цепей ДНК активных генов. В разных вариантах синдрома Кокейна могут иметь место нарушения функции нескольких генов (CSA, CSB, XPD и XPG), каждый из которых вносит свой вклад во взаимодействие процессов репарации и транскипции, как показано на рисунке выше (статья).
В разных вариантах синдрома Кокейна могут иметь место нарушения функции нескольких генов (CSA, CSB, XPD и XPG), каждый из которых вносит свой вклад во взаимодействие процессов репарации и транскипции, как показано на рисунке выше (статья).
Атаксия-телангиэктазия
Больные аутосомно-рецессивным заболеванием атаксией-телангиэктазией (Ataxia telangiectasia) страдают от нейрональной дегенерации, преждевременного старения и увеличения частоты возникновения опухолей. In vitro клетки больных данным синдромом ускоренно теряют теломеры вследствие их оксидативного повреждения. Пациенты с атаксией-телангиэктазией несут мутацию в гене Atm (ataxia teleangiectasia mutated), кодирующем киназу ATM, главный сенсор повреждения ДНК в клетке. Распознавая повреждение ДНК в контрольных точках клеточного цикла, ATM фосфорилирует такие белки-мишени как р53, Chk1, Chk2, BRCA1, NBS1, FANCD2, гистон h3AX, которые, в свою очередь, индуцируют задержку клеточного цикла и репарацию ДНК. Мутации в генах некоторых мишеней ATM тоже приводят к ускоренному старению.
Синдром поломок Ниджмеджена (NBS)
К таким мутациям относится и причина синдрома поломок Ниджмеджена (Nijmegen breakage syndrome). Больных отличает микроцефалия, особая форма лица, низкорослость, иммунодефицит, радиочувствительность, предрасположенность к лимфоидным разновидностям рака. В случае мутации гена NBS1 возникает нестабильность хромосом в результате дефекта структур Холлидея, образующихся в процессе пострепликативной рекомбинационной репарации двухцепочечных разрывов ДНК.
Анемия Фанкони
Симптомы анемии Фанкони (Fanconi anemia) — дефекты развития (например, отсутствие пальцев), нарушения функции красного костного мозга, острая миелогенная лейкемия и другие формы рака, зачастую не позволяющие дожить до зрелого возраста. Всего известно 7 генов, способных приводить к анемии Фанкони: FancA, FancB, FancC, FancD, FancE, FancF и FancG. Продукты этих генов фосфорилируются ATM и участвуют в репарации ДНК и задержке S-фазы клеточного цикла.
Связь прогерий с теломерами
Многие из перечисленных частичных прогерий сопряжены с короткими теломерами: синдром Вернера, RTS, прогерия Хатчинсона-Гилфорда, атаксия телангиэктазия, NBS. В свою очередь, ускоренное укорочение теломер при прогериях, обусловливая репликативное старение дифференцированных соматических клеток и дисфункцию стволовых клеток, вызывает симптомы, во многом напоминающие нормальное старение, такие как потеря волос, поседение, дистрофия ногтей, снижение костной массы, гематологические заболевания, иммунодефициты.
Врожденный дискератоз
Еще более убедительный пример роли теломер в прогериях и старении в целом — заболевание врожденный дискератоз (dyskeratosis congenita).
Врожденный дискератоз существует в двух формах. Х-сцепленная форма вызвана мутацией гена белка дискерина, участвующего в обеспечении функции теломеразы. Аутосомно-доминантная форма вызвана дефектом в гене РНК компонента теломеразы. В результате, пациенты с врожденным дискератозом имеют низкую активность теломеразы и короткие теломеры и умирают в ранней зрелости от нарушений в красном костном мозге, рака или легочных осложнений. Некоторые пациенты с врожденным дискератозом характеризуются высокой частотой карцином, что предполагает вклад укорочения теломер в возрастзависимое возникновение рака.
Некоторые пациенты с врожденным дискератозом характеризуются высокой частотой карцином, что предполагает вклад укорочения теломер в возрастзависимое возникновение рака.
Синдром Дауна
Мало кто знает, но к синдромам ускоренного старения относится и синдром Дауна (Down_syndrome). Синдром Дауна вызывается трисомией по 21-й хромосоме и среди его фенотипов — ранее начало возрастзависимых патологических изменений, а также укорочение продолжительности жизни. В мозге больных обнаруживаются активированные ферменты каспаза-3 и каспаза-8, обусловливающие апоптоз нейронов в тех участках, где происходит накопление -амилоида и нейрофибриллярных бляшек (белок-предшественник амилоида может быть потенциальным субстратом каспаз). Культивируемые in vitro нейроны мышей с трисомией 16-й хромосомы (сходной с 21-й хромосомой человека) имеют меньшую продолжительность жизни, чем нормальные фетальные нейроны и их гибель предотвращается ингибиторами каспаз (статья).
Общие причины прогерий
Как мы видим, разнообразие частичных прогерий огромно. В то же время, четко выявляются механизмы, их обусловливающие:
В то же время, четко выявляются механизмы, их обусловливающие:
— Нарушение свойств теломер, хроматина и клеточного ядра
— Нарушение репарации и репликации ДНК, генетическая нестабильность
— Нарушение экспрессии генов
— Репликативное старение
— Повышенная чувствительность клеток к апоптозу
— Элиминация стволовых клеток
Вполне вероятно, что те же самые механизмы задействованы в «нормальном» старении.
Перспективы исследований
Однако, следует признать, что не все еще ясно, например:
— В чем состоят сходства и в чем — различия частичных прогерий и обычного старения?
— Каким образом связаны между собой генетические причины всех прогерий, что является определяющим фактором?
— Какие из симптомов прогерий являются причиной, а какие — следствием ускоренного старения?
— Как противостоять ускоренному старению?
Исследовательские группы в области прогерий
Группа Паолы Скаффиди активно занимается исследованием молекулярных причин прогерии Хатчинсона-Гилфорда.
Сайт граппы:
http://ccr.cancer.gov/staff/staff.asp?profileid=6671
Фонд Мило Гладстейн по изучению синдрома Блума
Сайт фонда:
www.milogladsteinfoundation.org
Общество по изучению пигментной ксеродермы (XP Society)
Сайт общества:
www.xps.org
Группа Лерри Леба, изучающая молекулярную генетику синдрома Вернера.
Сайт группы:
http://depts.washington.edu/loeblabs/research/research.htm
Ссылки на полные тексты статей по теме
Наследственные синдромы с признаками преждевременного старения | Голоунина
1. López-Otín C, Blasco MA, Partridge L, et al. The Hallmarks of Aging. Cell. 2013;153(6):1194-1217. doi: https://doi.org/10.1016/j.cell.2013.05.039
2. Kudlow BA, Kennedy BK, Monnat RJ, Jr. Werner and Hutchinson-Gilford progeria syndromes: mechanistic basis of human progeroid diseases. Nat Rev Mol Cell Biol. 2007;8(5):394-404. doi: https://doi.org/10.1038/nrm2161
Werner and Hutchinson-Gilford progeria syndromes: mechanistic basis of human progeroid diseases. Nat Rev Mol Cell Biol. 2007;8(5):394-404. doi: https://doi.org/10.1038/nrm2161
3. Hutchinson J. Congenital Absence of Hair and Mammary Glands with Atrophic Condition of the Skin and its Appendages, in a Boy whose Mother had been almost wholly Bald from Alopecia Areata from the age of Six. Med Chir Trans. 1886;69:473-477. doi: https://doi.org/10.1177/095952878606900127
4. Gilford H. On a Condition of Mixed Premature and Immature Development. Med Chir Trans. 1897;80:17-46 25. doi: https://doi.org/10.1177/095952879708000105
5. Werner O. On cataract in conjunction with scleroderma. In: Salk D, Fujiwara Y, Martin GM, editors. Werner’s Syndrome and Human Aging. Advances in Experimental Medicine and Biology. Vol. 190. Boston: Springer; 1985. p. 1-14. doi: https://doi.org/10.1007/978-1-4684-7853-2_1.
Advances in Experimental Medicine and Biology. Vol. 190. Boston: Springer; 1985. p. 1-14. doi: https://doi.org/10.1007/978-1-4684-7853-2_1.
6. Rautenstrauch T, Snigula F. Progeria: a cell culture study and clinical report of familial incidence. Eur J Pediatr. 1977;124(2):101-111. doi: https://doi.org/10.1007/bf00477545
7. Wiedemann HR. An unidentified neonatal progeroid syndrome: follow-up report. Eur J Pediatr. 1979;130(1):65-70. doi: https://doi.org/10.1007/bf00441901
8. Wambach JA, Wegner DJ, Patni N, et al. Bi-allelic POLR3A Loss-of-Function Variants Cause Autosomal-Recessive Wiedemann-Rautenstrauch Syndrome. Am J Hum Genet. 2018;103(6):968-975. doi: https://doi.org/10.1016/j.ajhg.2018.10.010
9. Paolacci S, Li Y, Agolini E, et al. Specific combinations of biallelic POLR3A variants cause Wiedemann-Rautenstrauch syndrome. J Med Genet. 2018;55(12):837-846. doi: https://doi.org/10.1136/jmedgenet-2018-105528
Paolacci S, Li Y, Agolini E, et al. Specific combinations of biallelic POLR3A variants cause Wiedemann-Rautenstrauch syndrome. J Med Genet. 2018;55(12):837-846. doi: https://doi.org/10.1136/jmedgenet-2018-105528
10. Paolacci S, Bertola D, Franco J, et al. Wiedemann-Rautenstrauch syndrome: A phenotype analysis. Am J Med Genet A. 2017;173(7):1763-1772. doi: https://doi.org/10.1002/ajmg.a.38246
11. Becerra CH, Contreras-Garcia GA, Perez Vera LA, et al. Wiedemann-Rautenstrauch syndrome prenatal diagnosis. J Perinatol. 2014;34(12):954-956. doi: https://doi.org/10.1038/jp.2014.156
12. Beauregard-Lacroix E, Salian S, Kim H, et al. A variant of neonatal progeroid syndrome, or Wiedemann-Rautenstrauch syndrome, is associated with a nonsense variant in POLR3GL. Eur J Hum Genet. 2020;28(4):461-468. doi: https://doi.org/10.1038/s41431-019-0539-6
Eur J Hum Genet. 2020;28(4):461-468. doi: https://doi.org/10.1038/s41431-019-0539-6
13. Gargiuli C, Schena E, Mattioli E, et al. Lamins and bone disorders: current understanding and perspectives. Oncotarget. 2018;9(32):22817-22831. doi: https://doi.org/10.18632/oncotarget.25071
14. Дадали Е.Л., Билева Д.С., Угаров И.В. Клинико-генетическая характеристика наследственных ламинопатий. // Анналы клинической и экспериментальной неврологии. — 2008. — Т. 2. — №4. — С. 28-33. [Dadaly EL, Bileva DS, Ugarov IV. Clinical and genetic characteristics of hereditary laminopathies. Annaly klinicheskoy i eksperimental’noy nevrologii. 2008;2(4):28-33. (In Russ.)]
15. Gonzalo S, Kreienkamp R, Askjaer P. Hutchinson-Gilford Progeria Syndrome: A premature aging disease caused by LMNA gene mutations.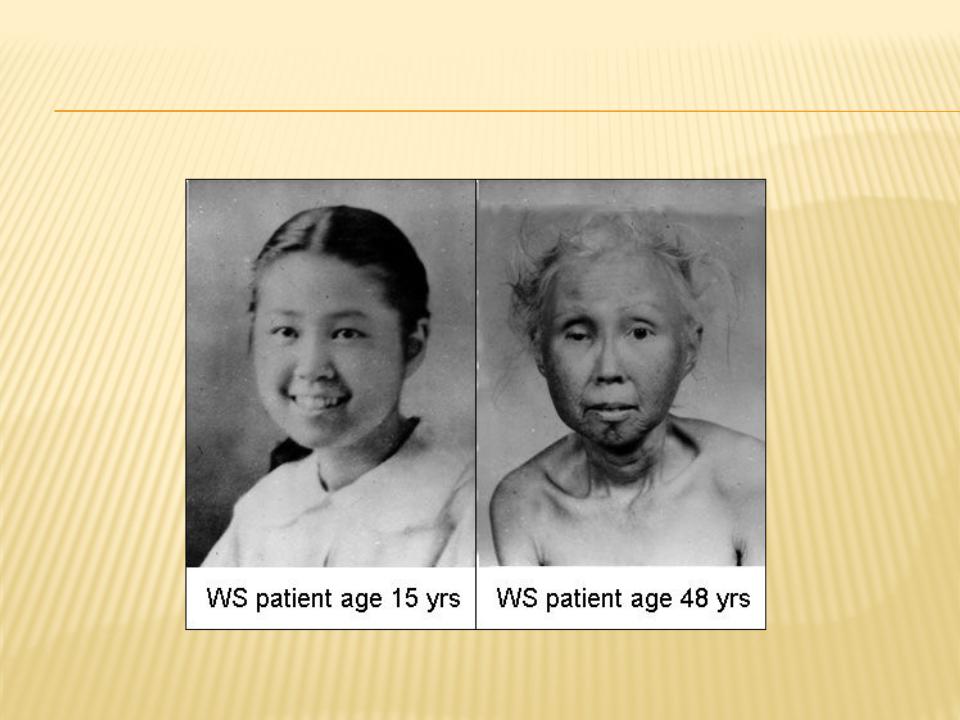 Ageing Res Rev. 2017;33:18-29. doi: https://doi.org/10.1016/j.arr.2016.06.007
Ageing Res Rev. 2017;33:18-29. doi: https://doi.org/10.1016/j.arr.2016.06.007
16. Ashapkin VV, Kutueva LI, Kurchashova SY, Kireev II. Are There Common Mechanisms Between the Hutchinson–Gilford Progeria Syndrome and Natural Aging? Front Genet. 2019;10. doi: https://doi.org/10.3389/fgene.2019.00455
17. Turgay Y, Eibauer M, Goldman AE, et al. The molecular architecture of lamins in somatic cells. Nature. 2017;543(7644):261-264. doi: https://doi.org/10.1038/nature21382
18. Лаврушкина С.В., Овсянникова Н.Л., Юдина А.С., и др. Канцерогенез и старение: взгляд со стороны ядерной ламины. // Цитология. — 2018. — Т. 60. — №11. — С. 892-894. [Lavrushlina SV, Ovsyannikova NL, Yudina AS, et al. Carcinogenesis and ageing: a view from nuclear lamina. Cell and tissue biology. 2018;60(11):892-894. (In Russ.)] doi: https://doi.org/10.1134/S0041377118110056.
Cell and tissue biology. 2018;60(11):892-894. (In Russ.)] doi: https://doi.org/10.1134/S0041377118110056.
19. Swahari V, Nakamura A. Speeding up the clock: The past, present and future of progeria. Dev Growth Differ. 2016;58(1):116-130. doi: https://doi.org/10.1111/dgd.12251
20. Piekarowicz K, Machowska M, Dzianisava V, Rzepecki R. Hutchinson-Gilford Progeria Syndrome—Current Status and Prospects for Gene Therapy Treatment. Cells. 2019;8(2):88. doi: https://doi.org/10.3390/cells8020088
21. Politano L, Lattanzi G, Benedetti S, et al. Emerging perspectives on laminopathies. Cell Health Cytoskelet. 2016:25. doi: https://doi.org/10.2147/chc.s59507
22. Hamczyk MR, del Campo L, Andrés V. Aging in the Cardiovascular System: Lessons from Hutchinson-Gilford Progeria Syndrome. Annu Rev Physiol. 2018;80(1):27-48. doi: https://doi.org/10.1146/annurev-physiol-021317-121454
Hamczyk MR, del Campo L, Andrés V. Aging in the Cardiovascular System: Lessons from Hutchinson-Gilford Progeria Syndrome. Annu Rev Physiol. 2018;80(1):27-48. doi: https://doi.org/10.1146/annurev-physiol-021317-121454
23. Navarro CL, Esteves-Vieira V, Courrier S, et al. New ZMPSTE24 (FACE1) mutations in patients affected with restrictive dermopathy or related progeroid syndromes and mutation update. Eur J Hum Genet. 2013;22(8):1002-1011. doi: https://doi.org/10.1038/ejhg.2013.258
24. McKenna T, Sola Carvajal A, Eriksson M. Skin Disease in Laminopathy-Associated Premature Aging. J Invest Dermatol. 2015;135(11):2577-2583. doi: https://doi.org/10.1038/jid.2015.295
25. Filesi I, Gullotta F, Lattanzi G, et al. Alterations of nuclear envelope and chromatin organization in mandibuloacral dysplasia, a rare form of laminopathy. Physiol Genomics. 2005;23(2):150-158. doi: https://doi.org/10.1152/physiolgenomics.00060.2005
Physiol Genomics. 2005;23(2):150-158. doi: https://doi.org/10.1152/physiolgenomics.00060.2005
26. Yaou RB, Navarro C, Quijano-Roy S, et al. Type B mandibuloacral dysplasia with congenital myopathy due to homozygous ZMPSTE24 missense mutation. Eur J Hum Genet. 2011;19(6):647-654. doi: https://doi.org/10.1038/ejhg.2010.256
27. Соркина Е.Л., Тюльпаков А.Н. Наследственные и приобретенные липодистрофии: молекулярно-генетические и аутоиммунные механизмы. // Ожирение и метаболизм. — 2018. — Т. 15. — №1. — С. 39-42. [Sorkina EL, Tyulpakov AN. Inherited and acquired lipodystrophies: molecular-genetic and autoimmune mechanisms. Obesity and metabolism. 2018;15(1):39-42. (In Russ.)] doi: https://doi.org/10.14341/OMET2018139-42
28. Bachrati CZ, Hickson ID. RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem J. 2003;374(Pt 3):577-606. doi: https://doi.org/10.1042/BJ20030491
RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem J. 2003;374(Pt 3):577-606. doi: https://doi.org/10.1042/BJ20030491
29. Croteau DL, Popuri V, Opresko PL, Bohr VA. Human RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem. 2014;83:519-552. doi: https://doi.org/10.1146/annurev-biochem-060713-035428
30. Guo RB, Rigolet P, Ren H, et al. Structural and functional analyses of disease-causing missense mutations in Bloom syndrome protein. Nucleic Acids Res. 2007;35(18):6297-6310. doi: https://doi.org/10.1093/nar/gkm536
31. Larizza L, Magnani I, Roversi G. Rothmund-Thomson syndrome and RECQL4 defect: splitting and lumping. Cancer Lett. 2006;232(1):107-120. doi: https://doi.org/10.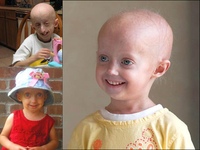 1016/j.canlet.2005.07.042
1016/j.canlet.2005.07.042
32. Shamanna RA, Croteau DL, Lee JH, Bohr VA. Recent Advances in Understanding Werner Syndrome. F1000Res. 2017;6:1779. doi: https://doi.org/10.12688/f1000research.12110.1
33. O’Sullivan RJ, Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol. 2010;11(3):171-181. doi: https://doi.org/10.1038/nrm2848
34. Johnson JE, Cao K, Ryvkin P, et al. Altered gene expression in the Werner and Bloom syndromes is associated with sequences having G-quadruplex forming potential. Nucleic Acids Res. 2010;38(4):1114-1122. doi: https://doi.org/10.1093/nar/gkp1103
35. Драпкина О.М., Шепель Р. Н. Теломеры и теломеразный комплекс. Основные клинические проявления генетического сбоя // Кардиоваскулярная терапия и профилактика. – 2015. Т.14. – №1. – С. 70-77. [Drapkina OM, Shepel RN. Telomeres and telomerase complex. The main clinical manifestation of genetic malfunctioning. Cardiovascular Therapy and Prevention 2015;14(1):70-77. (In Russ.)] doi: https://doi.org/10.15829/1728-8800-2015-1-70-77
Н. Теломеры и теломеразный комплекс. Основные клинические проявления генетического сбоя // Кардиоваскулярная терапия и профилактика. – 2015. Т.14. – №1. – С. 70-77. [Drapkina OM, Shepel RN. Telomeres and telomerase complex. The main clinical manifestation of genetic malfunctioning. Cardiovascular Therapy and Prevention 2015;14(1):70-77. (In Russ.)] doi: https://doi.org/10.15829/1728-8800-2015-1-70-77
36. Tang W, Robles AI, Beyer RP, et al. The Werner syndrome RECQ helicase targets G4 DNA in human cells to modulate transcription. Hum Mol Genet. 2016;25(10):2060-2069. doi: https://doi.org/10.1093/hmg/ddw079
37. Ishikawa N, Nakamura K-I, Izumiyama-Shimomura N, et al. Accelerated <i>in vivo</i> epidermal telomere loss in Werner syndrome. Aging. 2011;3(4):417-429. doi: https://doi.org/10.18632/aging.100315
38.![]() Maierhofer A, Flunkert J, Oshima J, et al. Accelerated epigenetic aging in Werner syndrome. Aging. 2017;9(4):1143-1152. doi: https://doi.org/10.18632/aging.101217
Maierhofer A, Flunkert J, Oshima J, et al. Accelerated epigenetic aging in Werner syndrome. Aging. 2017;9(4):1143-1152. doi: https://doi.org/10.18632/aging.101217
39. Zhang W, Li J, Suzuki K, et al. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science. 2015;348(6239):1160-1163. doi: https://doi.org/10.1126/science.aaa1356
40. Sarbacher CA, Halper JT. Connective Tissue and Age-Related Diseases. Subcell Biochem. 2019;91:281-310. doi: https://doi.org/10.1007/978-981-13-3681-2_11
41. Masala MV, Scapaticci S, Olivieri C, et al. Epidemiology and clinical aspects of Werner’s syndrome in North Sardinia: description of a cluster. Eur J Dermatol. 2007;17(3):213-216. doi: https://doi. org/10.1684/ejd.2007.0155
org/10.1684/ejd.2007.0155
42. Yokote K, Chanprasert S, Lee L, et al. WRN Mutation Update: Mutation Spectrum, Patient Registries, and Translational Prospects. Hum Mutat. 2017;38(1):7-15. doi: https://doi.org/10.1002/humu.23128
43. Nishimura EK, Granter SR, Fisher DE. Mechanisms of hair graying: incomplete melanocyte stem cell maintenance in the niche. Science. 2005;307(5710):720-724. doi: https://doi.org/10.1126/science.1099593
44. Oshima J, Sidorova JM, Monnat RJ, Jr. Werner syndrome: Clinical features, pathogenesis and potential therapeutic interventions. Ageing Res Rev. 2017;33:105-114. doi: https://doi.org/10.1016/j.arr.2016.03.002
45. Lessel D, Kubisch C. Hereditary Syndromes with Signs of Premature Aging. Dtsch Arztebl Int. 2019;116(29-30):489-496. doi: https://doi.org/10.3238/arztebl.2019.0489
Lessel D, Kubisch C. Hereditary Syndromes with Signs of Premature Aging. Dtsch Arztebl Int. 2019;116(29-30):489-496. doi: https://doi.org/10.3238/arztebl.2019.0489
46. Ozturk M, Akdeniz N, Ayakta H, Kosem M. A brother and sister with Werner’s syndrome demonstrating extensive tendon calcification and sacroiliitis. Clin Exp Dermatol. 2006;31(4):615-616. doi: https://doi.org/10.1111/j.1365-2230.2006.02130.x
47. Honjo S, Yokote K, Fujimoto M, et al. Clinical outcome and mechanism of soft tissue calcification in Werner syndrome. Rejuvenation Res. 2008;11(4):809-819. doi: https://doi.org/10.1089/rej.2007.0649
48. Leone A, Costantini AM, Brigida R, et al. Soft-tissue mineralization in Werner syndrome. Skeletal Radiol. 2005;34(1):47-51. doi: https://doi.org/10.1007/s00256-004-0792-8
doi: https://doi.org/10.1007/s00256-004-0792-8
49. Sickles CK, Gross GP. Progeria (Werner Syndrome). Treasure Island: StatPearls Publishing; 2020.
50. Belaya ZE, Grebennikova TA, Yashina JN, et al. Rare causes of secondary hyperparathyroidism clinical cases of Werner’s syndrome, Gitelman’s syndrome and osteopetrosis among patients referred for primary hyperparathyroidism. In: Osteoporosis International. Vol. 26. London: Springer; 2015. p. 111.
51. Lauper JM, Krause A, Vaughan TL, Monnat RJ, Jr. Spectrum and risk of neoplasia in Werner syndrome: a systematic review. PLoS One. 2013;8(4):e59709. doi: https://doi.org/10.1371/journal.pone.0059709
52. de Renty C, Ellis NA. Bloom’s syndrome: Why not premature aging?: A comparison of the BLM and WRN helicases. Ageing Res Rev. 2017;33:36-51. doi: https://doi.org/10.1016/j.arr.2016.05.010
de Renty C, Ellis NA. Bloom’s syndrome: Why not premature aging?: A comparison of the BLM and WRN helicases. Ageing Res Rev. 2017;33:36-51. doi: https://doi.org/10.1016/j.arr.2016.05.010
53. Bloom D. congenital telangiectatic erythema resembling lupus erythematosus in dwarfs<subtitle>Probably a Syndrome Entity. Arch Pediatr Adolesc Med. 1954;88(6):754. doi: https://doi.org/10.1001/archpedi.1954.02050100756008
54. German J, Sanz MM, Ciocci S, et al. Syndrome-causing mutations of the BLM gene in persons in the Bloom’s Syndrome Registry. Hum Mutat. 2007;28(8):743-753. doi: https://doi.org/10.1002/humu.20501
55. Fares F, Badarneh K, Abosaleh M, et al. Carrier frequency of autosomal-recessive disorders in the Ashkenazi Jewish population: should the rationale for mutation choice for screening be reevaluated? Prenat Diagn.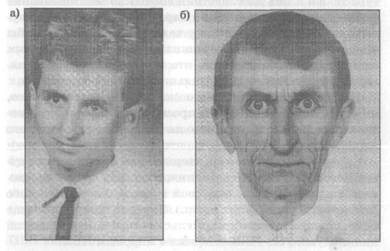 2008;28(3):236-241. doi: https://doi.org/10.1002/pd.1943
2008;28(3):236-241. doi: https://doi.org/10.1002/pd.1943
56. Kaneko H, Kondo N. Clinical features of Bloom syndrome and function of the causative gene, BLM helicase. Expert Rev Mol Diagn. 2004;4(3):393-401. doi: https://doi.org/10.1586/14737159.4.3.393
57. Lu L, Jin W, Wang LL. Aging in Rothmund-Thomson syndrome and related RECQL4 genetic disorders. Ageing Res Rev. 2017;33:30-35. doi: https://doi.org/10.1016/j.arr.2016.06.002
58. Thomson MS. Poikiloderma Congenitale: Two Cases for Diagnosis. Proc R Soc Med. 1936;29(5):453-455.
59. Colombo EA, Locatelli A, Cubells Sanchez L, et al. Rothmund-Thomson Syndrome: Insights from New Patients on the Genetic Variability Underpinning Clinical Presentation and Cancer Outcome. Int J Mol Sci. 2018;19(4). doi: https://doi.org/10.3390/ijms19041103
Int J Mol Sci. 2018;19(4). doi: https://doi.org/10.3390/ijms19041103
60. Oshima J, Kato H, Maezawa Y, Yokote K. RECQ helicase disease and related progeroid syndromes: RECQ2018 meeting. Mech Ageing Dev. 2018;173:80-83. doi: https://doi.org/10.1016/j.mad.2018.05.002
61. Hafsi W, Badri T. Poikiloderma Congenitale. Treasure Island (FL): StatPearls Publishing; 2019.
62. Araujo SJ, Kuraoka I. Nucleotide excision repair genes shaping embryonic development. Open Biol. 2019;9(10):190166. doi: https://doi.org/10.1098/rsob.190166
63. Spivak G. Nucleotide excision repair in humans. DNA Repair (Amst). 2015;36:13-18. doi: https://doi.org/10.1016/j. dnarep.2015.09.003
dnarep.2015.09.003
64. Moriwaki S, Kanda F, Hayashi M, et al. Xeroderma pigmentosum clinical practice guidelines. J Dermatol. 2017;44(10):1087-1096. doi: https://doi.org/10.1111/1346-8138.13907
65. Brooks BP, Thompson AH, Bishop RJ, et al. Ocular manifestations of xeroderma pigmentosum: long-term follow-up highlights the role of DNA repair in protection from sun damage. Ophthalmology. 2013;120(7):1324-1336. doi: https://doi.org/10.1016/j.ophtha.2012.12.044
66. Bradford PT, Goldstein AM, Tamura D, et al. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet. 2011;48(3):168-176. doi: https://doi.org/10.1136/jmg.2010.083022
67. Karass M, Naguib MM, Elawabdeh N, et al. Xeroderma pigmentosa: three new cases with an in depth review of the genetic and clinical characteristics of the disease. Fetal Pediatr Pathol. 2015;34(2):120-127. doi: https://doi.org/10.3109/15513815.2014.982336
Karass M, Naguib MM, Elawabdeh N, et al. Xeroderma pigmentosa: three new cases with an in depth review of the genetic and clinical characteristics of the disease. Fetal Pediatr Pathol. 2015;34(2):120-127. doi: https://doi.org/10.3109/15513815.2014.982336
68. Kaliki S, Jajapuram SD, Maniar A, Mishra DK. Ocular and Periocular Tumors in Xeroderma Pigmentosum: A Study of 120 Asian Indian Patients. Am J Ophthalmol. 2019;198:146-153. doi: https://doi.org/10.1016/j.ajo.2018.10.011
69. Black JO. Xeroderma Pigmentosum. Head Neck Pathol. 2016;10(2):139-144. doi: https://doi.org/10.1007/s12105-016-0707-8
70. Cockayne EA. Dwarfism with retinal atrophy and deafness. Arch Dis Child. 1936;11(61):1-8. doi: https://doi.org/10.1136/adc.11. 61.1
61.1
71. Wilson BT, Stark Z, Sutton RE, et al. The Cockayne Syndrome Natural History (CoSyNH) study: clinical findings in 102 individuals and recommendations for care. Genet Med. 2016;18(5):483-493. doi: https://doi.org/10.1038/gim.2015.110
72. Слижов П.А., Долинина Т.И., Плескай Н.М., и др. Маркеры старения в клетках больных синдромом Коккейна. Общие и индивидуальные различия. // Цитология. — 2018. — Т. 60. — №3. — С. 188-199. [Slizhov PA, Dolinina TI, Pleskach NM, et al. Aging markers in cells of patients with Cockayne Syndrome. General and individual differences. Cell and tissue biology. 2018;60(3):188-199. (In Russ.)] doi: https://doi.org/10.31116/tsitol.2018.03.05
73. Kubota M, Ohta S, Ando A, et al. Nationwide survey of Cockayne syndrome in Japan: Incidence, clinical course and prognosis. Pediatr Int. 2015;57(3):339-347. doi: https://doi.org/10.1111/ped.12635
Pediatr Int. 2015;57(3):339-347. doi: https://doi.org/10.1111/ped.12635
74. Karikkineth AC, Scheibye-Knudsen M, Fivenson E, et al. Cockayne syndrome: Clinical features, model systems and pathways. Ageing Res Rev. 2017;33:3-17. doi: https://doi.org/10.1016/j.arr.2016.08.002
75. Kalantaridou SN, Zoumakis E, Makrigiannakis A, et al. Corticotropin-releasing hormone, stress and human reproduction: an update. J Reprod Immunol. 2010;85(1):33-39. doi: https://doi.org/10.1016/j.jri.2010.02.005
76. Hayashi M, Miwa-Saito N, Tanuma N, Kubota M. Brain vascular changes in Cockayne syndrome. Neuropathology. 2012;32(2):113-117. doi: https://doi.org/10.1111/j.1440-1789.2011.01241.x
77. Kraemer KH, Patronas NJ, Schiffmann R, et al. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship. Neuroscience. 2007;145(4):1388-1396. doi: https://doi.org/10.1016/j.neuroscience.2006.12.020
Kraemer KH, Patronas NJ, Schiffmann R, et al. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship. Neuroscience. 2007;145(4):1388-1396. doi: https://doi.org/10.1016/j.neuroscience.2006.12.020
78. Pereira LB, Valente NYS, Rocha VB. Do you know this syndrome? Ichthyosis associated with neurological condition and alteration of hairs. An Bras Dermatol. 2018;93(1):135-137. doi: https://doi.org/10.1590/abd1806-4841.20187727
79. Farmaki E, Nedelkopoulou N, Delli F, et al. Brittle Hair, Photosensitivity, Brain Hypomyelination and Immunodeficiency: Clues to Trichothiodystrophy. Indian J Pediatr. 2017;84(1):89-90. doi: https://doi.org/10.1007/s12098-016-2209-9
80. Potter H, Chial HJ, Caneus J, et al. Chromosome Instability and Mosaic Aneuploidy in Neurodegenerative and Neurodevelopmental Disorders. Front Genet. 2019;10:1092. doi: https://doi.org/10.3389/fgene.2019.01092
Chromosome Instability and Mosaic Aneuploidy in Neurodegenerative and Neurodevelopmental Disorders. Front Genet. 2019;10:1092. doi: https://doi.org/10.3389/fgene.2019.01092
81. Khetarpal P, Das S, Panigrahi I, Munshi A. Primordial dwarfism: overview of clinical and genetic aspects. Mol Genet Genomics. 2016;291(1):1-15. doi: https://doi.org/10.1007/s00438-015-1110-y
82. O’Driscoll M, Ruiz-Perez VL, Woods CG, et al. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33(4):497-501. doi: https://doi.org/10.1038/ng1129
83. Qvist P, Huertas P, Jimeno S, et al. CtIP Mutations Cause Seckel and Jawad Syndromes. PLoS Genet. 2011;7(10):e1002310. doi: https://doi.org/10.1371/journal.pgen.1002310
doi: https://doi.org/10.1371/journal.pgen.1002310
84. Al-Dosari MS, Shaheen R, Colak D, Alkuraya FS. Novel CENPJ mutation causes Seckel syndrome. J Med Genet. 2010;47(6):411-414. doi: https://doi.org/10.1136/jmg.2009.076646
85. Kalay E, Yigit G, Aslan Y, et al. CEP152 is a genome maintenance protein disrupted in Seckel syndrome. Nat Genet. 2011;43(1):23-26. doi: https://doi.org/10.1038/ng.725
86. Sir JH, Barr AR, Nicholas AK, et al. A primary microcephaly protein complex forms a ring around parental centrioles. Nat Genet. 2011;43(11):1147-1153. doi: https://doi.org/10.1038/ng.971
87. Dauber A, Lafranchi SH, Maliga Z, et al. Novel microcephalic primordial dwarfism disorder associated with variants in the centrosomal protein ninein. J Clin Endocrinol Metab. 2012;97(11):E2140-2151. doi: https://doi.org/10.1210/jc.2012-2150
J Clin Endocrinol Metab. 2012;97(11):E2140-2151. doi: https://doi.org/10.1210/jc.2012-2150
88. Shaheen R, Faqeih E, Ansari S, et al. Genomic analysis of primordial dwarfism reveals novel disease genes. Genome Res. 2014;24(2):291-299. doi: https://doi.org/10.1101/gr.160572.113
89. Ogi T, Walker S, Stiff T, et al. Identification of the first ATRIP-deficient patient and novel mutations in ATR define a clinical spectrum for ATR-ATRIP Seckel Syndrome. PLoS Genet. 2012;8(11):e1002945. doi: https://doi.org/10.1371/journal.pgen.1002945
90. Barbelanne M, Tsang WY. Molecular and cellular basis of autosomal recessive primary microcephaly. Biomed Res Int. 2014;2014:547986. doi: https://doi.org/10.1155/2014/547986
91. Savage SA. Dyskeratosis Congenita. In: Adam MP, Ardinger HH, Pagon RA, et al, editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993-2020.
Savage SA. Dyskeratosis Congenita. In: Adam MP, Ardinger HH, Pagon RA, et al, editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993-2020.
92. Sharma RK, Gupta M, Sood S, Gupta A. Dyskeratosis congenita: presentation of cutaneous triad in a sporadic case. BMJ Case Rep. 2018;11(1). doi: https://doi.org/10.1136/bcr-2018-226736
93. Aplas V. Poikiloderma, parapsoriasis and atrophia cutis cum pigmentatione, dystrophia ungium et leukoplakia oris Zinsser, so-called dyskeratosis congenita. Arch Klin Exp Dermatol. 1956;202(3):224-237. doi: https://doi.org/10.1007/bf00476707
94. Wang F, Du YQ, Gong W, et al. Research progress of dyskeratosis congenita. Zhonghua Kou Qiang Yi Xue Za Zhi. 2019;54(2):130-134. doi: https://doi. org/10.3760/cma.j.issn.1002-0098.2019.02.010
95. Savage SA, Bertuch AA. The genetics and clinical manifestations of telomere biology disorders. Genet Med. 2010;12(12):753-764. doi: https://doi.org/10.1097/GIM.0b013e3181f415b5
96. Dodson LM, Baldan A, Nissbeck M, et al. From incomplete penetrance with normal telomere length to severe disease and telomere shortening in a family with monoallelic and biallelic PARN pathogenic variants. Hum Mutat. 2019;40(12):2414-2429. doi: https://doi.org/10.1002/humu.23898
97. Savage SA. Beginning at the ends: telomeres and human disease. F1000Res. 2018;7. doi: https://doi.org/10.12688/f1000research.14068.1
98. Kutbay NO, Yurekli BS, Erdemir Z, et al. A case of dyskeratosis congenita associated with hypothyroidism and hypogonadism. Hormones (Athens). 2016;15(2):297-299. doi: https://doi.org/10.14310/horm.2002.1655
Kutbay NO, Yurekli BS, Erdemir Z, et al. A case of dyskeratosis congenita associated with hypothyroidism and hypogonadism. Hormones (Athens). 2016;15(2):297-299. doi: https://doi.org/10.14310/horm.2002.1655
99. Shomali W, Brar R. Late presentation of dyskeratosis congenita. Br J Haematol. 2019;187(3):273. doi: https://doi.org/10.1111/bjh.16131
100. Du H, Guo Y, Ma D, et al. A case report of heterozygous TINF2 gene mutation associated with pulmonary fibrosis in a patient with dyskeratosis congenita. Medicine (Baltimore). 2018;97(19):e0724. doi: https://doi.org/10.1097/MD.0000000000010724
101. de Boer J, Andressoo JO, de Wit J, et al. Premature aging in mice deficient in DNA repair and transcription. Science. 2002;296(5571):1276-1279. doi: https://doi.org/10.1126/science.1070174
doi: https://doi.org/10.1126/science.1070174
102. Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochim Biophys Acta. 2007;1775(1):138-162. doi: https://doi.org/10.1016/j.bbcan.2006.08.007
103. Zhang W, Li J, Suzuki K, et al. Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science. 2015;348(6239):1160-1163. doi: https://doi.org/10.1126/science.aaa1356
104. Shumaker DK, Dechat T, Kohlmaier A, et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A. 2006;103(23):8703-8708. doi: https://doi.org/10.1073/pnas.0602569103
105. Davis T, Brook AJ, Rokicki MJ, et al. Evaluating the Role of p38 MAPK in the Accelerated Cell Senescence of Werner Syndrome Fibroblasts. Pharmaceuticals (Basel). 2016;9(2). doi: https://doi.org/10.3390/ph9020023
Davis T, Brook AJ, Rokicki MJ, et al. Evaluating the Role of p38 MAPK in the Accelerated Cell Senescence of Werner Syndrome Fibroblasts. Pharmaceuticals (Basel). 2016;9(2). doi: https://doi.org/10.3390/ph9020023
106. Tivey HS, Brook AJ, Rokicki MJ, et al. p38 (MAPK) stress signalling in replicative senescence in fibroblasts from progeroid and genomic instability syndromes. Biogerontology. 2013;14(1):47-62. doi: https://doi.org/10.1007/s10522-012-9407-2
107. Bagley MC, Davis T, Murziani PG, et al. Use of p38 MAPK Inhibitors for the Treatment of Werner Syndrome. Pharmaceuticals (Basel). 2010;3(6):1842-1872. doi: https://doi.org/10.3390/ph4061842
108. Yamaga M, Takemoto M, Shoji M, et al. Werner syndrome: a model for sarcopenia due to accelerated aging. Aging (Albany NY). 2017;9(7):1738-1744. doi: https://doi.org/10.18632/aging.101265
Aging (Albany NY). 2017;9(7):1738-1744. doi: https://doi.org/10.18632/aging.101265
109. von Walden F, Liu C, Aurigemma N, Nader GA. mTOR signaling regulates myotube hypertrophy by modulating protein synthesis, rDNA transcription, and chromatin remodeling. Am J Physiol Cell Physiol. 2016;311(4):C663-C672. doi: https://doi.org/10.1152/ajpcell.00144.2016
110. Dormond O. mTOR in Human Diseases. Int J Mol Sci. 2019;20(9). doi: https://doi.org/10.3390/ijms20092351
111. Ou HL, Schumacher B. DNA damage responses and p53 in the aging process. Blood. 2018;131(5):488-495. doi: https://doi.org/10.1182/blood-2017-07-746396
112. Wu D, Prives C. Relevance of the p53-MDM2 axis to aging.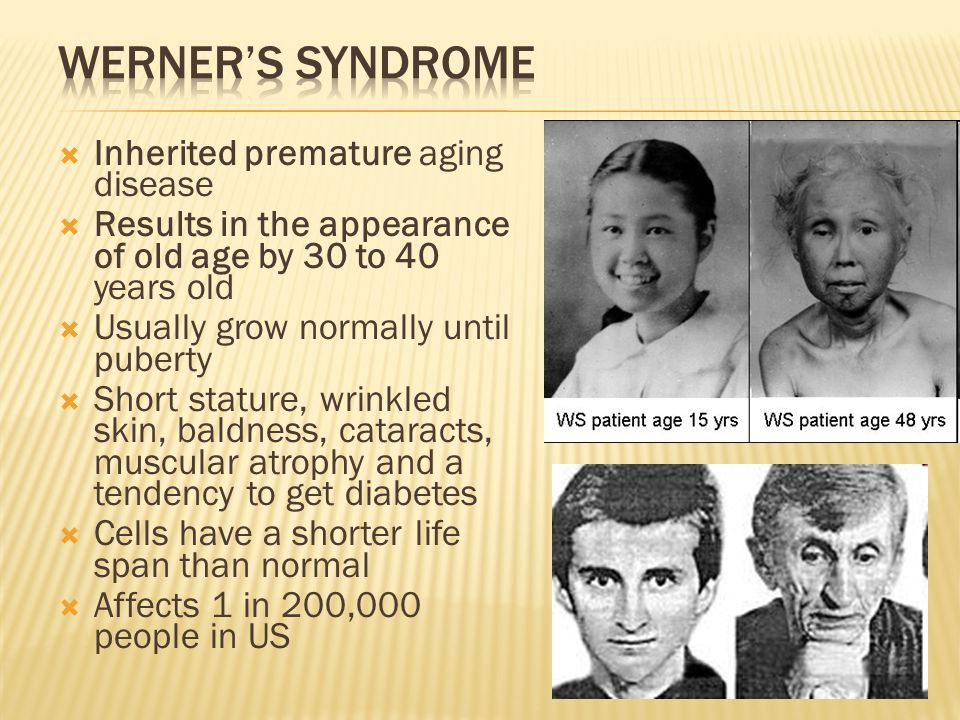 Cell Death Differ. 2018;25(1):169-179. doi: https://doi.org/10.1038/cdd.2017.187
Cell Death Differ. 2018;25(1):169-179. doi: https://doi.org/10.1038/cdd.2017.187
113. Inoki K, Ouyang H, Li Y, Guan KL. Signaling by target of rapamycin proteins in cell growth control. Microbiol Mol Biol Rev. 2005;69(1):79-100. doi: https://doi.org/10.1128/MMBR.69.1.79-100.2005
114. Weichhart T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology. 2018;64(2):127-134. doi: https://doi.org/10.1159/000484629
115. Wiza C, Nascimento EB, Ouwens DM. Role of PRAS40 in Akt and mTOR signaling in health and disease. Am J Physiol Endocrinol Metab. 2012;302(12):E1453-1460. doi: https://doi.org/10.1152/ajpendo.00660.2011
116. Shaw RJ, Bardeesy N, Manning BD, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6(1):91-99. doi: https://doi.org/10.1016/j.ccr.2004.06.007
Shaw RJ, Bardeesy N, Manning BD, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6(1):91-99. doi: https://doi.org/10.1016/j.ccr.2004.06.007
117. Mammucari C, Milan G, Romanello V, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6(6):458-471. doi: https://doi.org/10.1016/j.cmet.2007.11.001
118. Zhao J, Brault JJ, Schild A, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6(6):472-483. doi: https://doi.org/10.1016/j.cmet.2007.11.004
119. Wu JJ, Liu J, Chen EB, et al. Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mTOR expression. Cell Rep. 2013;4(5):913-920. doi: https://doi.org/10.1016/j.celrep.2013.07.030
Cell Rep. 2013;4(5):913-920. doi: https://doi.org/10.1016/j.celrep.2013.07.030
120. Vellai T, Takacs-Vellai K, Zhang Y, et al. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003;426(6967):620. doi: https://doi.org/10.1038/426620a
121. Bjedov I, Toivonen JM, Kerr F, et al. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010;11(1):35-46. doi: https://doi.org/10.1016/j.cmet.2009.11.010
122. Kaeberlein M, Powers RW, 3rd, Steffen KK, et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310(5751):1193-1196. doi: https://doi.org/10.1126/science.1115535
123. Seto B. Rapamycin and mTOR: a serendipitous discovery and implications for breast cancer. Clin Transl Med. 2012;1(1):29. doi: https://doi.org/10.1186/2001-1326-1-29
Seto B. Rapamycin and mTOR: a serendipitous discovery and implications for breast cancer. Clin Transl Med. 2012;1(1):29. doi: https://doi.org/10.1186/2001-1326-1-29
124. Demidenko ZN, Zubova SG, Bukreeva EI, et al. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8(12):1888-1895. doi: https://doi.org/10.4161/cc.8.12.8606
125. Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460(7253):392-395. doi: https://doi.org/10.1038/nature08221
126. Oral EA, Simha V, Ruiz E, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346(8):570-578. doi: https://doi.org/10.1056/NEJMoa012437
127. Brown RJ, Oral EA, Cochran E, et al. Long-term effectiveness and safety of metreleptin in the treatment of patients with generalized lipodystrophy. Endocrine. 2018;60(3):479-489. doi: https://doi.org/10.1007/s12020-018-1589-1
128. Gordon LB, Kleinman ME, Miller DT, et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2012;109(41):16666-16671. doi: https://doi.org/10.1073/pnas.1202529109
129. Gordon LB, Massaro J, D’Agostino RB, Sr., et al. Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation. 2014;130(1):27-34. doi: https://doi.org/10.1161/CIRCULATIONAHA.113.008285
130. Gordon LB, Shappell H, Massaro J, et al. Association of Lonafarnib Treatment vs No Treatment With Mortality Rate in Patients With Hutchinson-Gilford Progeria Syndrome. JAMA. 2018;319(16):1687-1695. doi: https://doi.org/10.1001/jama.2018.3264
Association of Lonafarnib Treatment vs No Treatment With Mortality Rate in Patients With Hutchinson-Gilford Progeria Syndrome. JAMA. 2018;319(16):1687-1695. doi: https://doi.org/10.1001/jama.2018.3264
131. McNally EM, Wyatt EJ. Welcome to the splice age: antisense oligonucleotide-mediated exon skipping gains wider applicability. J Clin Invest. 2016;126(4):1236-1238. doi: https://doi.org/10.1172/JCI86799
132. Lee JM, Nobumori C, Tu Y, et al. Modulation of LMNA splicing as a strategy to treat prelamin A diseases. J Clin Invest. 2016;126(4):1592-1602. doi: https://doi.org/10.1172/JCI85908
133. Ершова О.Б., Белова К.Ю., Дегтярев А.А., и др. Анализ летальности у пациентов с переломом проксимального отдела бедра. // Остеопороз и остеопатии.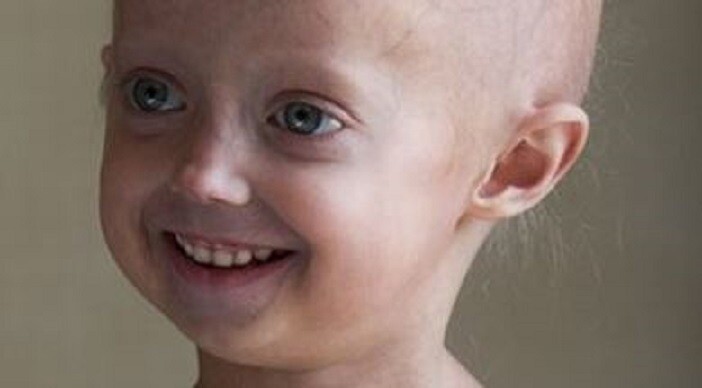 — 2015. — Т. 18. — №3. — С. 3-8. [Ershova OB, Belova KY, Degtyarev AA, et al. Analysis of mortality in patients with a fracture of the proximal femur. Osteoporosis and bone diseases. 2015;18(3):3-8. (In Russ.)] doi: https://doi.org/10.14341/osteo201533-8
— 2015. — Т. 18. — №3. — С. 3-8. [Ershova OB, Belova KY, Degtyarev AA, et al. Analysis of mortality in patients with a fracture of the proximal femur. Osteoporosis and bone diseases. 2015;18(3):3-8. (In Russ.)] doi: https://doi.org/10.14341/osteo201533-8
134. Мельниченко Г.А., Белая Ж.Е., Рожинская Л.Я., и др. Краткое изложение клинических рекомендаций по диагностике и лечению остеопороза Российской ассоциации эндокринологов. // Остеопороз и остеопатии. — 2016. — Т. 19. — №3. — С. 28-36. [Melnichenko GA, Belaya ZE, Rozhinskaya LY, et al. Summary of clinical guidelines for the diagnosis and treatment of osteoporosis of the Russian association of endocrinologists. Osteoporosis and bone diseases 2016;19(3):28-36. (In Russ.)] doi: https://doi.org/10.14341/osteo2016328-36
135. Белая Ж.Е., Рожинская Л.Я. Витамин D в терапии остеопороза: его роль в комбинации с препаратами для лечения остеопороза, внескелетные эффекты. // Эффективная фармакотерапия. — 2013. — Т. 38. — №2. — С. 14-29. [Belaya ZY, Rozhinskaya LY. Vitamin D in the treatment of osteoporosis: its role in the combination with antiosteoporotic therapy, non-skeletal effects. Effektivnaya farmakoterapiya. 2013;38(2):14-29. (In Russ.)]
// Эффективная фармакотерапия. — 2013. — Т. 38. — №2. — С. 14-29. [Belaya ZY, Rozhinskaya LY. Vitamin D in the treatment of osteoporosis: its role in the combination with antiosteoporotic therapy, non-skeletal effects. Effektivnaya farmakoterapiya. 2013;38(2):14-29. (In Russ.)]
136. Geusens PP, Lems WF. Fracture prevention in postmenopausal women with osteoporosis by an annual infusion of zoledronic acid. Ned Tijdschr Geneeskd. 2007;151(26):1445-1448.
137. Lyles KW, Colon-Emeric CS, Magaziner JS, et al. Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med. 2007;357(18):1799-1809. doi: https://doi.org/10.1056/NEJMoa074941
138. Cummings SR, Lui LY, Eastell R, Allen IE. Association Between Drug Treatments for Patients With Osteoporosis and Overall Mortality Rates: A Meta-analysis. JAMA Intern Med. 2019. doi: https://doi.org/10.1001/jamainternmed.2019.2779
JAMA Intern Med. 2019. doi: https://doi.org/10.1001/jamainternmed.2019.2779
139. Bliuc D, Tran T, van Geel T, et al. Mortality risk reduction differs according to bisphosphonate class: a 15-year observational study. Osteoporos Int. 2019;30(4):817-828. doi: https://doi.org/10.1007/s00198-018-4806-0
140. Lee P, Ng C, Slattery A, et al. Preadmission Bisphosphonate and Mortality in Critically Ill Patients. J Clin Endocrinol Metab. 2016;101(5):1945-1953. doi: https://doi.org/10.1210/jc.2015-3467
141. Bergman J, Nordstrom A, Hommel A, et al. Bisphosphonates and mortality: confounding in observational studies? Osteoporos Int. 2019;30(10):1973-1982. doi: https://doi.org/10.1007/s00198-019-05097-1
142. Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA. Metformin as a Tool to Target Aging. Cell Metab. 2016;23(6):1060-1065. doi: https://doi.org/10.1016/j.cmet.2016.05.011
Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA. Metformin as a Tool to Target Aging. Cell Metab. 2016;23(6):1060-1065. doi: https://doi.org/10.1016/j.cmet.2016.05.011
143. Anisimov VN, Berstein LM, Egormin PA, et al. Metformin slows down aging and extends life span of female SHR mice. Cell Cycle. 2008;7(17):2769-2773. doi: https://doi.org/10.4161/cc.7.17.6625
144. Landman GW, Kleefstra N, van Hateren KJ, et al. Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care. 2010;33(2):322-326. doi: https://doi.org/10.2337/dc09-1380
145. Lee MS, Hsu CC, Wahlqvist ML, et al. Type 2 diabetes increases and metformin reduces total, colorectal, liver and pancreatic cancer incidences in Taiwanese: a representative population prospective cohort study of 800,000 individuals. BMC Cancer. 2011;11:20. doi: https://doi.org/10.1186/1471-2407-11-20
BMC Cancer. 2011;11:20. doi: https://doi.org/10.1186/1471-2407-11-20
146. Tseng CH. Diabetes, metformin use, and colon cancer: a population-based cohort study in Taiwan. Eur J Endocrinol. 2012;167(3):409-416. doi: https://doi.org/10.1530/EJE-12-0369
147. Tosca L, Rame C, Chabrolle C, et al. Metformin decreases IGF1-induced cell proliferation and protein synthesis through AMP-activated protein kinase in cultured bovine granulosa cells. Reproduction. 2010;139(2):409-418. doi: https://doi.org/10.1530/REP-09-0351
148. Karnevi E, Said K, Andersson R, Rosendahl AH. Metformin-mediated growth inhibition involves suppression of the IGF-I receptor signalling pathway in human pancreatic cancer cells. BMC Cancer. 2013;13:235. doi: https://doi. org/10.1186/1471-2407-13-235
org/10.1186/1471-2407-13-235
149. Zi FM, He JS, Li Y, et al. Metformin displays anti-myeloma activity and synergistic effect with dexamethasone in in vitro and in vivo xenograft models. Cancer Lett. 2015;356(2 Pt B):443-453. doi: https://doi.org/10.1016/j.canlet.2014.09.050
150. Niehr F, von Euw E, Attar N, et al. Combination therapy with vemurafenib (PLX4032/RG7204) and metformin in melanoma cell lines with distinct driver mutations. J Transl Med. 2011;9:76. doi: https://doi.org/10.1186/1479-5876-9-76
151. Colquhoun AJ, Venier NA, Vandersluis AD, et al. Metformin enhances the antiproliferative and apoptotic effect of bicalutamide in prostate cancer. Prostate Cancer Prostatic Dis. 2012;15(4):346-352. doi: https://doi.org/10.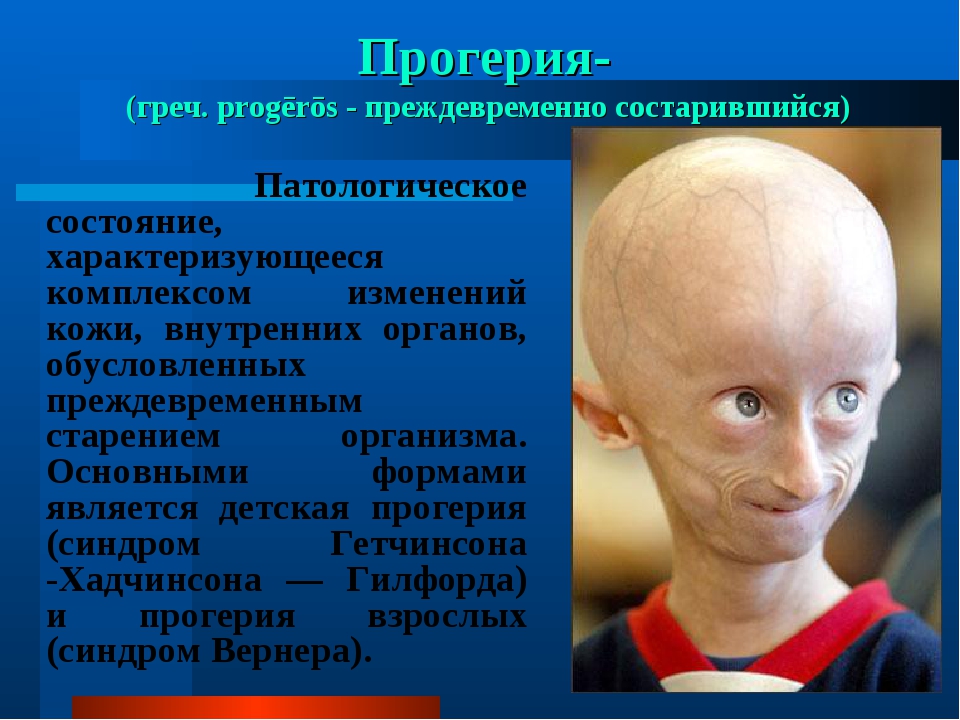 1038/pcan.2012.16
1038/pcan.2012.16
152. Li L, Han R, Xiao H, et al. Metformin sensitizes EGFR-TKI-resistant human lung cancer cells in vitro and in vivo through inhibition of IL-6 signaling and EMT reversal. Clin Cancer Res. 2014;20(10):2714-2726. doi: https://doi.org/10.1158/1078-0432.CCR-13-2613
153. Blandino G, Valerio M, Cioce M, et al. Metformin elicits anticancer effects through the sequential modulation of DICER and c-MYC. Nat Commun. 2012;3:865. doi: https://doi.org/10.1038/ncomms1859
154. Xu Y, Lu S. Metformin inhibits esophagus cancer proliferation through upregulation of USP7. Cell Physiol Biochem. 2013;32(5):1178-1186. doi: https://doi.org/10.1159/000354517
155. Algire C, Amrein L, Zakikhani M, et al. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr Relat Cancer. 2010;17(2):351-360. doi: https://doi.org/10.1677/erc-09-0252
Algire C, Amrein L, Zakikhani M, et al. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr Relat Cancer. 2010;17(2):351-360. doi: https://doi.org/10.1677/erc-09-0252
156. Gandini S, Puntoni M, Heckman-Stoddard BM, et al. Metformin and Cancer Risk and Mortality: A Systematic Review and Meta-analysis Taking into Account Biases and Confounders. Cancer Prev Res (Phila). 2014;7(9):867-885. doi: https://doi.org/10.1158/1940-6207.capr-13-0424
157. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet. 1998;352(9131):854-865. doi: https://doi.org/10.1016/s0140-6736(98)07037-8
158. Lautrup S, Caponio D, Cheung H-H, et al. Studying Werner syndrome to elucidate mechanisms and therapeutics of human aging and age-related diseases. Biogerontology. 2019;20(3):255-269. doi: https://doi.org/10.1007/s10522-019-09798-2
Lautrup S, Caponio D, Cheung H-H, et al. Studying Werner syndrome to elucidate mechanisms and therapeutics of human aging and age-related diseases. Biogerontology. 2019;20(3):255-269. doi: https://doi.org/10.1007/s10522-019-09798-2
Как диагноз влияет на жизнь — Wonderzine
По словам Виктории, она не сталкивалась с травлей по поводу своей внешности и никогда не воспринимала её как повод для расстройства: «Мою маму воспитали глухонемые родители, и я считаю это гораздо более удивительной историей. Конечно, бывало, кто-то кричал мне вслед гадости — но это единичные случаи, которые не привели к комплексам. Многие мои подруги — красивые девочки, у которых модельная внешность. Бывает, они начинают рассказывать мне о своих комплексах, о „неидеальном“ теле, о том, что не могут найти себе пару — меня это искренне поражает. Я не сталкивалась никогда с такими проблемами. Я приняла и полюбила себя такой, какая я есть. А то, чем была не особенно довольна, постаралась изменить».
А то, чем была не особенно довольна, постаралась изменить».
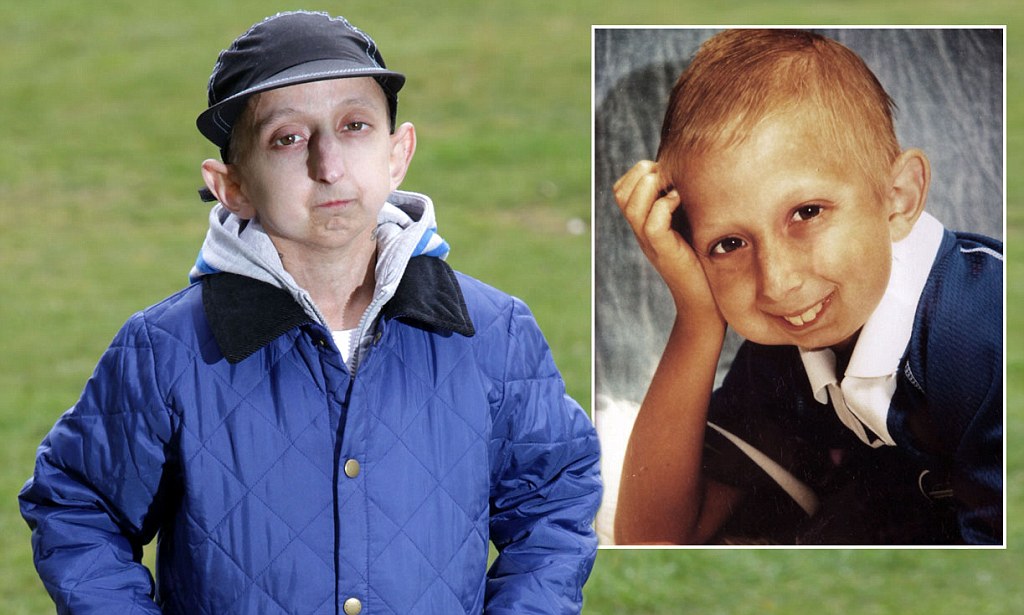
К сожалению, не всем, кто столкнулся с ускоренным старением, легко принять себя, да и социум этому не способствует: люди с прогерией регулярно попадают в зачем-то составляемые рейтинги «самых уродливых в мире», а истории об удачном применении новых видов терапии выходят с заголовками вроде «Врачи спасли девушку-старушку». Француженка Тифенн рассказывает, что самое сложное в этой болезни — взгляды окружающих и тот факт, что у тебя, по сути, нет детства и юности, ты непреднамеренно оказываешься в мире взрослых. «В детстве меня дразнили старухой, бабушкой, ведьмой… С возрастом шутки стали тоньше, но не перестали быть обидными. Не уверена, что люди понимают, как больно они могут сделать своими вопросами».
Из-за того, что синдромы преждевременного старения встречаются редко, о них плохо осведомлены и медицинские работники. По словам той же Тифенн, всё детство её направляли от одного врача к другому, но не могли сказать ничего определённого. На сайте ассоциации Cutis Laxa Internationale многие рассказывают, что их детям долго не могли поставить диагноз, а о существовании других людей с этим состоянием они даже не подозревали. Ассоциация создала закрытую группу поддержки для людей с эластозом. Изучением этого заболевания активно занимаются в Университете Питтсбурга — понимание мутаций, которые могут быть его причиной, поможет разработать диагностические методы и потенциально лечение.
На сайте ассоциации Cutis Laxa Internationale многие рассказывают, что их детям долго не могли поставить диагноз, а о существовании других людей с этим состоянием они даже не подозревали. Ассоциация создала закрытую группу поддержки для людей с эластозом. Изучением этого заболевания активно занимаются в Университете Питтсбурга — понимание мутаций, которые могут быть его причиной, поможет разработать диагностические методы и потенциально лечение.
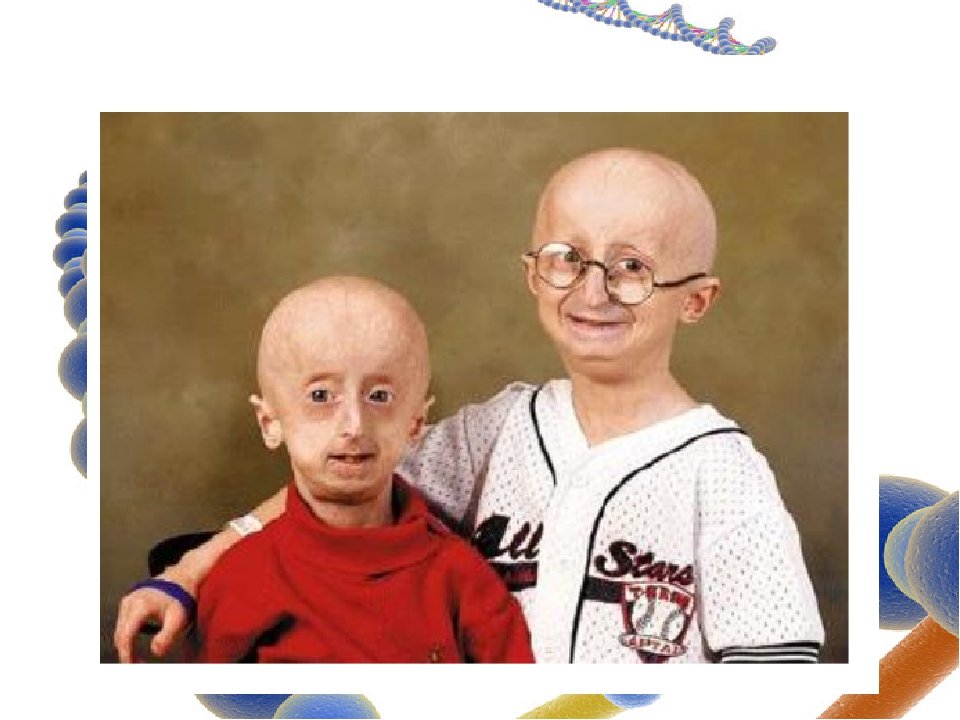
На 31 марта 2019 года в мире насчитывалось 157 детей с прогерией. Фонд изучения прогерии — главная организация, которая занимается этим вопросом. Фонд был основан в 1999 году родителями мальчика по имени Сэм, столкнувшимися с полным отсутствием информации по теме. В буклете фонда для родителей и тех, кто ухаживает за детьми с этим состоянием, отдельная глава посвящена тому, как вести себя с ребёнком и справляться с чужими взглядами, вопросами и комментариями — как отмечает один из родителей, «с самого начала нас больше волновала не медицинская сторона вопроса, а психологический и эмоциональный комфорт нашего ребёнка». Помимо множества технических рекомендаций по уходу за глазами, кожей, полостью рта, акцент делается на том, что интеллект у детей с прогерией соответствует возрасту, а почки, печень, желудочно-кишечный тракт и иммунитет работают нормально.
Помимо множества технических рекомендаций по уходу за глазами, кожей, полостью рта, акцент делается на том, что интеллект у детей с прогерией соответствует возрасту, а почки, печень, желудочно-кишечный тракт и иммунитет работают нормально.
В Европе поддержкой пациентов и их семей занимается организация Progeria Family Circle. В 2011 году вышла книга Хейли Окинс, девушки с прогерией, которой на тот момент было тринадцать лет. На сайте TED можно найти выступление Сэма, того самого мальчика, родители которого основали Фонд изучения прогерии, записанное в октябре 2013 года, за три месяца до его смерти.
ФОТОГРАФИИ: exopixel — stock.adobe.com
Расстройства, связанные с ускоренным старением — Проблемы со здоровьем у пожилых людей
Прогероидные синдромы — это редкие расстройства, вызывающие преждевременное старение и укорачивающие ожидаемую продолжительность жизни.
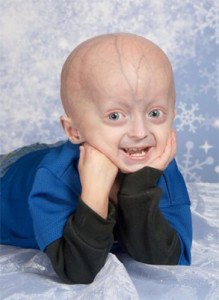
При прогероидных синдромах процесс старения значительно ускоряется. У пораженных детей развиваются все внешние признаки старения, включая облысение, сутулость, сухость и неэластичность кожи и морщины. Однако, в отличие от нормального старения, яичники или яички не работают, что приводит к стерильности. У девочек отсутствуют менструации. Пораженные такими синдромами дети имеют необычно низкий рост. Поэтому прогероидные синдромы не являются точной моделью ускоренного старения.
У пораженных детей развиваются все внешние признаки старения, включая облысение, сутулость, сухость и неэластичность кожи и морщины. Однако, в отличие от нормального старения, яичники или яички не работают, что приводит к стерильности. У девочек отсутствуют менструации. Пораженные такими синдромами дети имеют необычно низкий рост. Поэтому прогероидные синдромы не являются точной моделью ускоренного старения.
Существует несколько прогероидных синдромов. При синдроме Хатчинсона-Гилфорда и синдроме Вернера центральная нервная система, а, следовательно, и способность заниматься многими повседневными занятиями остаются в основном незатронутыми, если только не произойдет инсульт.
Синдром Хатчинсона-Гилфорда начинается в раннем детстве. Его причиной является генетическая аномалия, но обычно он не передается по наследству. Это значит, что генетическая аномалия (мутация) возникает сама по себе. Она вызывает снижение эластичности кожи и образование морщин, облысение и другие проблемы, обычно связанные со старением (например, заболевания сердца, почек и легких, а также остеопороз). Тело не может нормально расти, и поэтому кажется слишком маленьким по сравнению с головой. Большинство детей с синдромом Хатчинсона-Гилфорда умирают в подростковом возрасте. Причиной смерти обычно является сердечный приступ или инфаркт миокарда. Мутация, вызывающая это расстройство, была выявлена, и клиническое исследование с применением ингибитора дефектного гена принесло многообещающие результаты.
Тело не может нормально расти, и поэтому кажется слишком маленьким по сравнению с головой. Большинство детей с синдромом Хатчинсона-Гилфорда умирают в подростковом возрасте. Причиной смерти обычно является сердечный приступ или инфаркт миокарда. Мутация, вызывающая это расстройство, была выявлена, и клиническое исследование с применением ингибитора дефектного гена принесло многообещающие результаты.
Синдром Вернера (наследственный синдром) начинает развиваться в подростковом или раннем взрослом возрасте. Это вызывает снижение эластичности кожи и образование морщин, облысение и проблемы, связанные со старением, в том числе:
Синдром Вернера, ген RECQL2 м.
Исследуемый материал Цельная кровь (с ЭДТА)
Метод определения
Секвенирование. Выдаётся заключение врача-генетика!
Выдаётся заключение врача-генетика!
Исследование мутаций в гене RECQL2.
Тип наследования.
Аутосомно-рецессивный.
Гены, ответственные за развитие заболевания.
Ген RECQL2 (RECQ PROTEIN-LIKE 2) расположен на хромосоме 8 в регионе 8p12. Содержит 35 экзонов.
Определение заболевания.
Характеризуется симптомами преждевременного старения, возникающими после завершения полового созревания.
Патогенез и клиническая картина.
Молекулярно-генетической причиной заболевания являются мутации в гене RECQL2. Роль продуцируемого им белка WRN — поддержание геномной стабильности. WRN выполняет функцию геликазы, участвует в процессах репарации и репликации.
WRN выполняет функцию геликазы, участвует в процессах репарации и репликации.
Первые признаки синдрома Вернера могут возникать в возрасте 14-18 лет, проявляясь отставанием в росте, поседением, которое быстро становится универсальным и иногда сочетается с прогрессирующей алопецией. Обычно же синдром Вернера развивается после 20 лет, сопровождается преждевременным облысением, бледностью и истончением кожи конечностей и лица, которая становится резко натянутой, и под ней четко прослеживается сеть поверхностных кровеносных сосудов; подкожная жировая клетчатка и подлежащие мыщцы подвергаются атрофии, в результате чего конечности становятся непропорционально тонкими. Кожа над костными выступами постепенно утолщается и изъязвляется.
На 3-м десятилетии синдрома Вернера возникают двусторонняя катаракта, изменения голоса (слабый, хриплый и высокий), а также поражения кожи: склероцермоподобные изменения лица и конечностей, язвы на ногах, сухость кожи, мозоли в области подошв, телеангиэктазии. Больные приобретают характерный вид: низкий рост, лунообразное лицо с заостренными чертами, «птичьим носом», резко выступающим подбородком, сужением ротового отверстия (лицо имеет сходство со «склеродермической маской»), псеводоэкзофтальм, полное туловище и тонкие конечности. Атрофия сальных и потовых желез приводит к снижению сало- и потоотделения. На костных выступах и дистальных отделах конечностей возникают очаги гиперкератоза, отмечается диффузная гиперпигментация с участками гипопигментации; на стопах и голенях после травм образуются трофические язвы, на подошвах — кератоз; изменяются ногтевые пластинки. Помимо истончения и атрофии конечностей типичны костно-мышечные изменения, метастатическая кальцификация, генерализованный остеопороз, эрозивные остеоартриты, ограничение подвижности пальцев кистей (напоминающие склеродактилию), сгибательные контрактуры, болезненность конечностей, плоскостопие, деформации кистей (подобные таковым при ревматоидном артрите), осложненный септический артрит, остеомиелит.
Больные приобретают характерный вид: низкий рост, лунообразное лицо с заостренными чертами, «птичьим носом», резко выступающим подбородком, сужением ротового отверстия (лицо имеет сходство со «склеродермической маской»), псеводоэкзофтальм, полное туловище и тонкие конечности. Атрофия сальных и потовых желез приводит к снижению сало- и потоотделения. На костных выступах и дистальных отделах конечностей возникают очаги гиперкератоза, отмечается диффузная гиперпигментация с участками гипопигментации; на стопах и голенях после травм образуются трофические язвы, на подошвах — кератоз; изменяются ногтевые пластинки. Помимо истончения и атрофии конечностей типичны костно-мышечные изменения, метастатическая кальцификация, генерализованный остеопороз, эрозивные остеоартриты, ограничение подвижности пальцев кистей (напоминающие склеродактилию), сгибательные контрактуры, болезненность конечностей, плоскостопие, деформации кистей (подобные таковым при ревматоидном артрите), осложненный септический артрит, остеомиелит. При рентгенологическом исследовании обнаруживаются остеопороз костей голеней и стоп, метастатические гетеротопические кальцинаты мягких тканей (кожи, подкожной жировой клетчатки и др.), особенно сухожилий и связок коленных, локтевых и голеностопных суставов и периартикулярных тканей. Отмечаются медленно прогрессирующая катаракта, преждевременный атеросклероз с нарушением деятельности сердечно-сосудистой системы. У многих больных снижен интеллект.
При рентгенологическом исследовании обнаруживаются остеопороз костей голеней и стоп, метастатические гетеротопические кальцинаты мягких тканей (кожи, подкожной жировой клетчатки и др.), особенно сухожилий и связок коленных, локтевых и голеностопных суставов и периартикулярных тканей. Отмечаются медленно прогрессирующая катаракта, преждевременный атеросклероз с нарушением деятельности сердечно-сосудистой системы. У многих больных снижен интеллект.
На 4-м десятилетии синдрома Вернера, нередко на фоне эндокринных заболеваний (сахарного диабета, гипогонадизма, дисфункции паращитовидных желез и др.), у 5-10% больных развиваются злокачественные новообразования внутренних органов, костей (рак молочной железы, тиреоидная аденокарцинома, остеогенная саркома, менингиома, астроцитома и т.д), кожные раки. Смерть обычно наступает от сердечно-сосудистых заболеваний и злокачественных новообразований.
Для больных характерны бесплодие, импотенция, гинекомастия. При синдроме Вернера наблюдается генетически детерминированная нестабильность хромосом. Этот феномен выражается в постоянном, возникающем спонтанно или под действием некоторых агентов, изменении структуры хромосом, их отдельных локусов или групп локусов. Исследование фибробластов больных показало уменьшение потенциала делений в культуре клеток (они исчерпывают свой пролиферативный потенциал в 3-5 раз быстрее, чем клетки здоровых доноров) и 10-кратное увеличение темпа спонтанных мутаций, а также быстрое снижение длины теломерных повторов.
Этот феномен выражается в постоянном, возникающем спонтанно или под действием некоторых агентов, изменении структуры хромосом, их отдельных локусов или групп локусов. Исследование фибробластов больных показало уменьшение потенциала делений в культуре клеток (они исчерпывают свой пролиферативный потенциал в 3-5 раз быстрее, чем клетки здоровых доноров) и 10-кратное увеличение темпа спонтанных мутаций, а также быстрое снижение длины теломерных повторов.
Частота встречаемости: не установлена. Заболевание редкое.
Перечень исследуемых мутаций может быть предоставлен по запросу.
Литература
- Козлова С. И., Демикова Н. С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, 2007 – 448 с.
- Кеннет Л. Джонс «Наследственные синдромы по Девиду Смиту» Атлас-справочник.
 Москва, Практика, 2011.
Москва, Практика, 2011. - Галеева Н.М., Чухрова А.Л., Тверская С.М., Поляков А.В. «Новая мутация (р.М3871I) в гене RECQL2 у пациентов с синдромом Вернера»//Сб. материалов конференции «Генетика человека и патология», Томск 2007,144.
- Фёдорова Е. В. О врождённой прогерии. — 1980. — Т. 4. — С. 66. — (Педиатрия).
- Epstein, C. J., Martin, G. M., Schultz, A. L., Motulsky, A. G. Werner’s syndrome: a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine 45: 177-222, 1966.
- Moser, M. J., Oshima, J., Monnat, R. J., Jr. WRN mutations in Werner syndrome. Hum. Mutat. 13: 271-279, 1999. Note: Erratum: Hum. Mutat. 14: 84-85, 1999.
- OMIM.
 Москва, Практика, 2011.
Москва, Практика, 2011. Каково это – жить с преждевременным старением — Истории на TJ
Нобуаки Нагашима страдает редким синдромом, который ускоряет процесс старения. И надеется однажды помочь найти лекарство.
И надеется однажды помочь найти лекарство.
{«id»:98851,»url»:»https:\/\/tjournal.ru\/stories\/98851-kakovo-eto-zhit-s-prezhdevremennym-stareniem»,»title»:»\u041a\u0430\u043a\u043e\u0432\u043e \u044d\u0442\u043e \u2013 \u0436\u0438\u0442\u044c \u0441 \u043f\u0440\u0435\u0436\u0434\u0435\u0432\u0440\u0435\u043c\u0435\u043d\u043d\u044b\u043c \u0441\u0442\u0430\u0440\u0435\u043d\u0438\u0435\u043c»,»services»:{«vkontakte»:{«url»:»https:\/\/vk.com\/share.php?url=https:\/\/tjournal.ru\/stories\/98851-kakovo-eto-zhit-s-prezhdevremennym-stareniem&title=\u041a\u0430\u043a\u043e\u0432\u043e \u044d\u0442\u043e \u2013 \u0436\u0438\u0442\u044c \u0441 \u043f\u0440\u0435\u0436\u0434\u0435\u0432\u0440\u0435\u043c\u0435\u043d\u043d\u044b\u043c \u0441\u0442\u0430\u0440\u0435\u043d\u0438\u0435\u043c»,»short_name»:»VK»,»title»:»\u0412\u041a\u043e\u043d\u0442\u0430\u043a\u0442\u0435″,»width»:600,»height»:450},»facebook»:{«url»:»https:\/\/www. facebook.com\/sharer\/sharer.php?u=https:\/\/tjournal.ru\/stories\/98851-kakovo-eto-zhit-s-prezhdevremennym-stareniem»,»short_name»:»FB»,»title»:»Facebook»,»width»:600,»height»:450},»twitter»:{«url»:»https:\/\/twitter.com\/intent\/tweet?url=https:\/\/tjournal.ru\/stories\/98851-kakovo-eto-zhit-s-prezhdevremennym-stareniem&text=\u041a\u0430\u043a\u043e\u0432\u043e \u044d\u0442\u043e \u2013 \u0436\u0438\u0442\u044c \u0441 \u043f\u0440\u0435\u0436\u0434\u0435\u0432\u0440\u0435\u043c\u0435\u043d\u043d\u044b\u043c \u0441\u0442\u0430\u0440\u0435\u043d\u0438\u0435\u043c»,»short_name»:»TW»,»title»:»Twitter»,»width»:600,»height»:450},»telegram»:{«url»:»tg:\/\/msg_url?url=https:\/\/tjournal.ru\/stories\/98851-kakovo-eto-zhit-s-prezhdevremennym-stareniem&text=\u041a\u0430\u043a\u043e\u0432\u043e \u044d\u0442\u043e \u2013 \u0436\u0438\u0442\u044c \u0441 \u043f\u0440\u0435\u0436\u0434\u0435\u0432\u0440\u0435\u043c\u0435\u043d\u043d\u044b\u043c \u0441\u0442\u0430\u0440\u0435\u043d\u0438\u0435\u043c»,»short_name»:»TG»,»title»:»Telegram»,»width»:600,»height»:450},»odnoklassniki»:{«url»:»http:\/\/connect.
facebook.com\/sharer\/sharer.php?u=https:\/\/tjournal.ru\/stories\/98851-kakovo-eto-zhit-s-prezhdevremennym-stareniem»,»short_name»:»FB»,»title»:»Facebook»,»width»:600,»height»:450},»twitter»:{«url»:»https:\/\/twitter.com\/intent\/tweet?url=https:\/\/tjournal.ru\/stories\/98851-kakovo-eto-zhit-s-prezhdevremennym-stareniem&text=\u041a\u0430\u043a\u043e\u0432\u043e \u044d\u0442\u043e \u2013 \u0436\u0438\u0442\u044c \u0441 \u043f\u0440\u0435\u0436\u0434\u0435\u0432\u0440\u0435\u043c\u0435\u043d\u043d\u044b\u043c \u0441\u0442\u0430\u0440\u0435\u043d\u0438\u0435\u043c»,»short_name»:»TW»,»title»:»Twitter»,»width»:600,»height»:450},»telegram»:{«url»:»tg:\/\/msg_url?url=https:\/\/tjournal.ru\/stories\/98851-kakovo-eto-zhit-s-prezhdevremennym-stareniem&text=\u041a\u0430\u043a\u043e\u0432\u043e \u044d\u0442\u043e \u2013 \u0436\u0438\u0442\u044c \u0441 \u043f\u0440\u0435\u0436\u0434\u0435\u0432\u0440\u0435\u043c\u0435\u043d\u043d\u044b\u043c \u0441\u0442\u0430\u0440\u0435\u043d\u0438\u0435\u043c»,»short_name»:»TG»,»title»:»Telegram»,»width»:600,»height»:450},»odnoklassniki»:{«url»:»http:\/\/connect.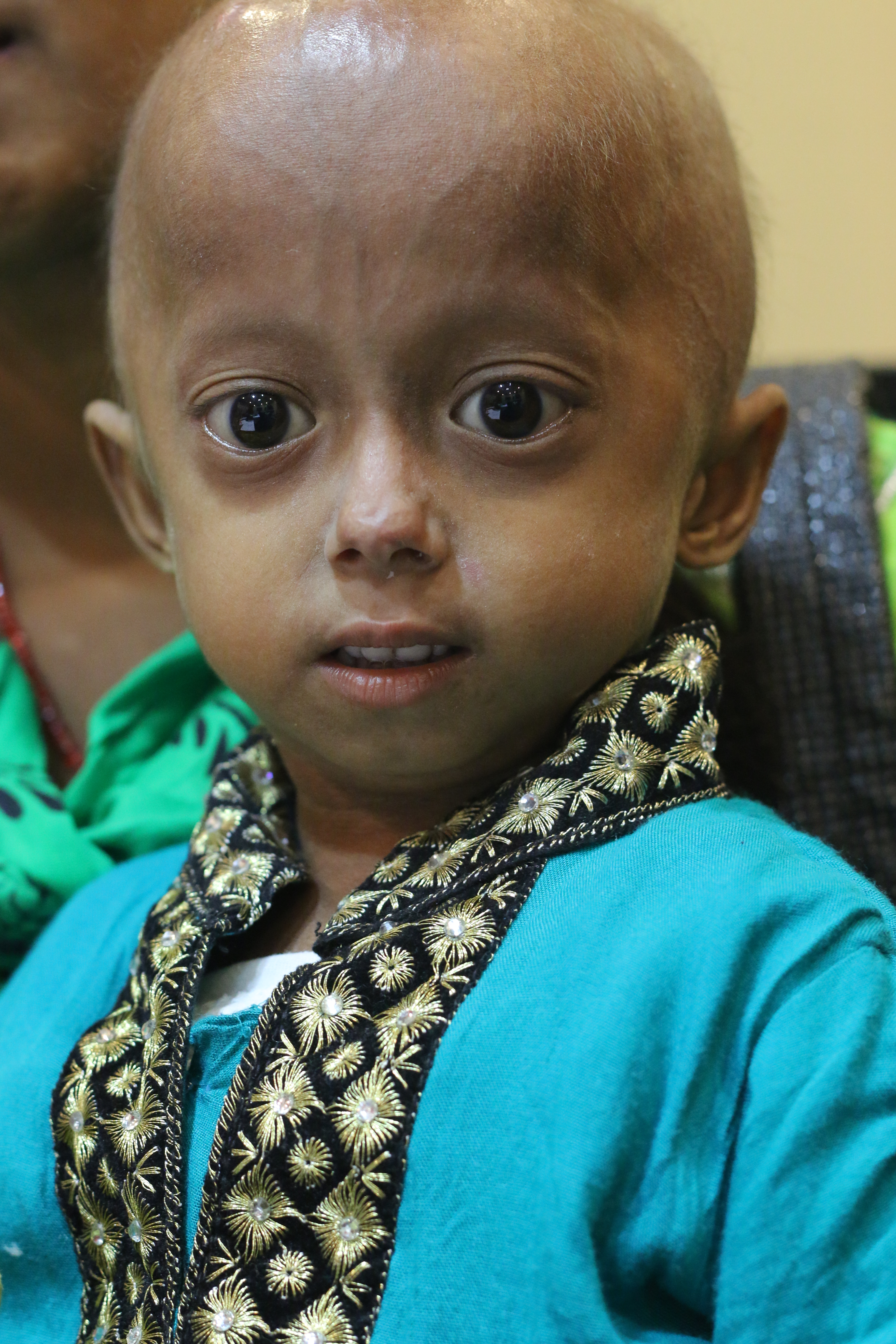 ok.ru\/dk?st.cmd=WidgetSharePreview&service=odnoklassniki&st.shareUrl=https:\/\/tjournal.ru\/stories\/98851-kakovo-eto-zhit-s-prezhdevremennym-stareniem»,»short_name»:»OK»,»title»:»\u041e\u0434\u043d\u043e\u043a\u043b\u0430\u0441\u0441\u043d\u0438\u043a\u0438″,»width»:600,»height»:450},»email»:{«url»:»mailto:?subject=\u041a\u0430\u043a\u043e\u0432\u043e \u044d\u0442\u043e \u2013 \u0436\u0438\u0442\u044c \u0441 \u043f\u0440\u0435\u0436\u0434\u0435\u0432\u0440\u0435\u043c\u0435\u043d\u043d\u044b\u043c \u0441\u0442\u0430\u0440\u0435\u043d\u0438\u0435\u043c&body=https:\/\/tjournal.ru\/stories\/98851-kakovo-eto-zhit-s-prezhdevremennym-stareniem»,»short_name»:»Email»,»title»:»\u041e\u0442\u043f\u0440\u0430\u0432\u0438\u0442\u044c \u043d\u0430 \u043f\u043e\u0447\u0442\u0443″,»width»:600,»height»:450}},»isFavorited»:false}
ok.ru\/dk?st.cmd=WidgetSharePreview&service=odnoklassniki&st.shareUrl=https:\/\/tjournal.ru\/stories\/98851-kakovo-eto-zhit-s-prezhdevremennym-stareniem»,»short_name»:»OK»,»title»:»\u041e\u0434\u043d\u043e\u043a\u043b\u0430\u0441\u0441\u043d\u0438\u043a\u0438″,»width»:600,»height»:450},»email»:{«url»:»mailto:?subject=\u041a\u0430\u043a\u043e\u0432\u043e \u044d\u0442\u043e \u2013 \u0436\u0438\u0442\u044c \u0441 \u043f\u0440\u0435\u0436\u0434\u0435\u0432\u0440\u0435\u043c\u0435\u043d\u043d\u044b\u043c \u0441\u0442\u0430\u0440\u0435\u043d\u0438\u0435\u043c&body=https:\/\/tjournal.ru\/stories\/98851-kakovo-eto-zhit-s-prezhdevremennym-stareniem»,»short_name»:»Email»,»title»:»\u041e\u0442\u043f\u0440\u0430\u0432\u0438\u0442\u044c \u043d\u0430 \u043f\u043e\u0447\u0442\u0443″,»width»:600,»height»:450}},»isFavorited»:false}
31 991 просмотров
Мужчина в японском метро Фото AFP
Когда уроженцу японского северного острова Хоккайдо Нобуаки Нагашиме (Nobuaki Nagashima) было около 25 лет, он стал чувствовать, будто его тело разваливается.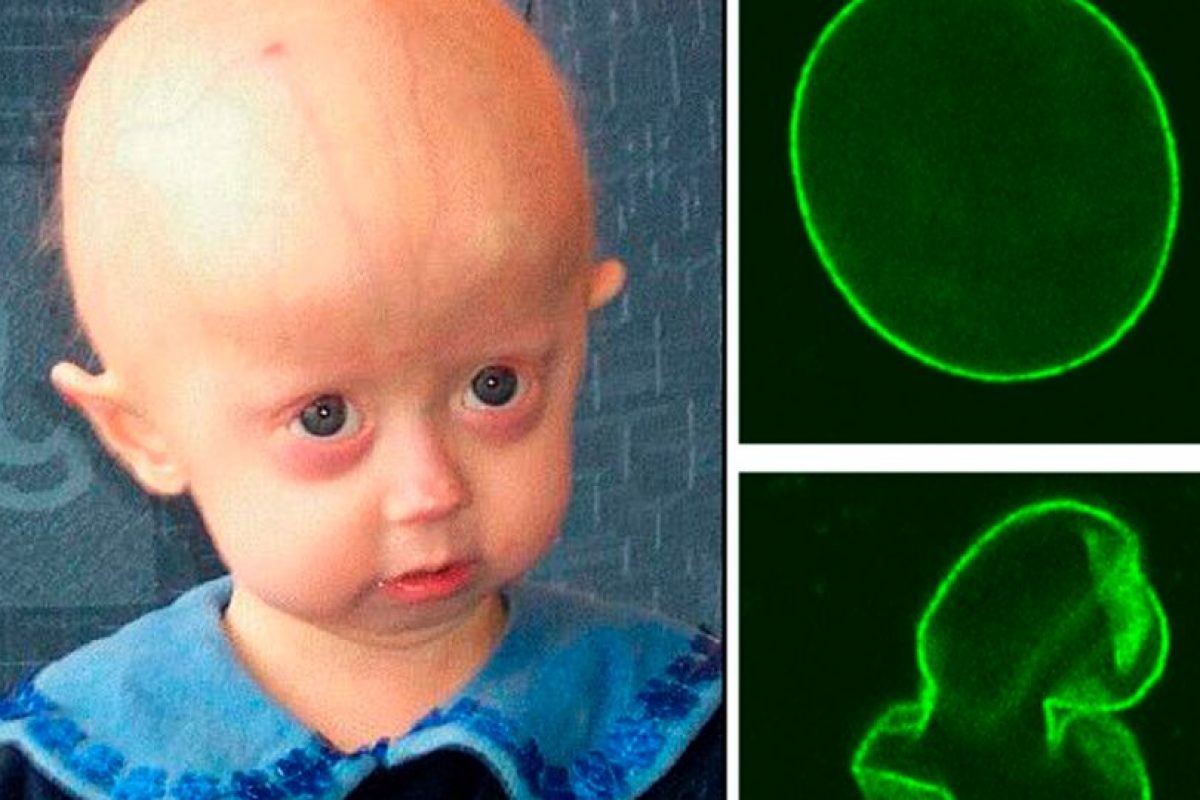 Объяснить это было невозможно – он 12 лет тренировался в армии, оттачивая навыки в любое время года (не уточняется, как он оказался в армии в юном возрасте). Однако тело сдавалось: в 25 лет у него обнаружили катаракту (расстройство зрения с угрозой полной его утраты), в 28 стали регулярно болеть бедра, а к 30 годам начала разрушаться кожа на ногах.
Объяснить это было невозможно – он 12 лет тренировался в армии, оттачивая навыки в любое время года (не уточняется, как он оказался в армии в юном возрасте). Однако тело сдавалось: в 25 лет у него обнаружили катаракту (расстройство зрения с угрозой полной его утраты), в 28 стали регулярно болеть бедра, а к 30 годам начала разрушаться кожа на ногах.
Через три года Нагашиме диагностировали синдром Вернера, который характеризуется преждевременным старением с широким спектром патологий. Включая диабет, атеросклероз, доброкачественные и злокачественные опухоли.
Лекарства от недуга не существует, так что десять лет спустя, к 43 годам, некогда крепкий Нагашиме превратился в дряхлого старика. О том, как устроен этот синдром, и может ли он в будущем помочь победить старость, рассказывает научное издание Mosaic. В материале не приводятся фотографии жертв недуга.
В материале не приводятся фотографии жертв недуга.
Старость в 25 лет
Последние 10 лет Нагашиме стал частым гостем во всех различных больницах. Болезнь лишила его возможности продолжить военную карьеру и вынудила пройти через пять-шесть операций по всему телу. С тех пор, как ему поставили диагноз, он похудел на 15 килограммов, не может пройти без трости больше нескольких метров и способен лишь подрабатывать в городской ратуше, когда у него есть силы.
Когда он впервые узнал о своей болезни, то не сдерживал слез и не скрывал диагноз от родителей. Услышав новость, его мать попросила прощения, что не могла родить более здорового человека.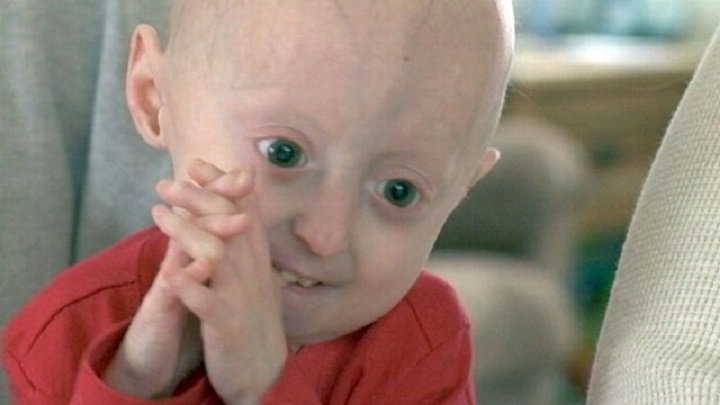
Отец Нагашимы сказал, что если он может принять и терпеть болезнь, то он по-настоящему сильный человек, и однажды, возможно, поможет учёным добыть знания, необходимые для спасения других людей.
Помимо X и Y половых хромосом, человек наследует две копии каждого гена в наших телах – от матери и отца. Синдром Вернера – это так называемое аутосомно-рецессивное расстройство. Это означает, что он проявляется только тогда, когда человек наследует мутировавшую версию гена WRN от обоих родителей.
Родители Нагашимы стареют нормально. У каждого из них рабочая копия гена WRN, так что их тела не показывают никаких признаков болезни.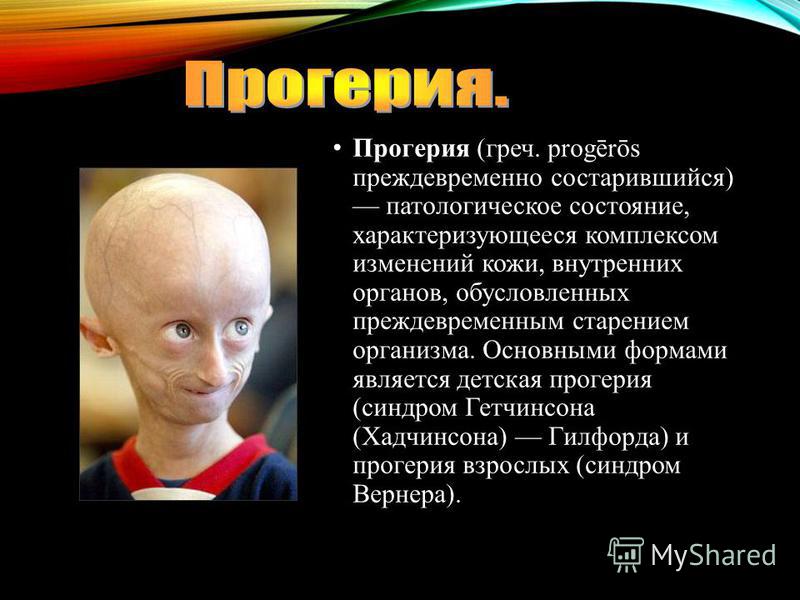 Аналогичная ситуация с его дедушкой и бабушкой, которым около 90 лет. Но сам японец получил две копии мутировавшего гена WRN. Ранее, по крайней мере, достоверно, никто из его семьи с этим не сталкивался.
Аналогичная ситуация с его дедушкой и бабушкой, которым около 90 лет. Но сам японец получил две копии мутировавшего гена WRN. Ранее, по крайней мере, достоверно, никто из его семьи с этим не сталкивался.
Этот ген открыли только в 1996 году, и с тех пор фиксировали незначительное число проявлений синдрома. По данным на 2008 год (более свежей информации нет), в мире задокументировано лишь 1487 случаев, 1128 из которых в Японии. Хотя такая статистика указывает, что это уникальное явление Японии, руководитель Международного реестра синдрома Вернера в Вашингтонском университете Джордж Мартин считает, что число реальных случаев примерно в семь раз превышает официальные данные. О большинстве врачи просто не знают.
Дисбаланс японских случаев объясняется двумя факторами. Во-первых, местные горы и острова создают изолирующий эффект, который на протяжении всей истории оказывал влияние на население – люди в более отдалённых регионах в прошлом с большей вероятностью заводили детей с кем-то генетически похожим на себя. Аналогичный эффект на итальянском острове Сардиния, где также фиксировали группу с синдромом Вернера. Во-вторых, развитость японской медицинской системы помогает эффективнее выявлять редкий недуг, чем в других странах.
Во-первых, местные горы и острова создают изолирующий эффект, который на протяжении всей истории оказывал влияние на население – люди в более отдалённых регионах в прошлом с большей вероятностью заводили детей с кем-то генетически похожим на себя. Аналогичный эффект на итальянском острове Сардиния, где также фиксировали группу с синдромом Вернера. Во-вторых, развитость японской медицинской системы помогает эффективнее выявлять редкий недуг, чем в других странах.
В университетской больнице в Тибе хранятся записи о 269 диагностированных пациентах, 116 из которых ещё живы. Одна из них – Сачи Суда (Sachi Suga), которая передвигается только на инвалидной коляске. Её мышцы настолько слабы, что она не может залезть или вылезти из ванны самостоятельно, а у плиты может стоять не больше одной-двух минут.
Неспособность приготовить еду она замещает супом быстрого приготовления, а затем, примерно в 5:30 утра, уходит на работу. Три раза в неделю её навещает социальный работник, который помогает ей справиться с болью в ногах. «Порой боль такая сильная, что лучше бы ноги просто отрезали», — говорит женщина.
Три раза в неделю её навещает социальный работник, который помогает ей справиться с болью в ногах. «Порой боль такая сильная, что лучше бы ноги просто отрезали», — говорит женщина.
Единственный плюс её состояния –в свои 64 года она уже перешла среднюю ожидаемую продолжительность жизни (до 55 лет) людей, страдающих синдромом Вернера.
С недавних пор в университетской больнице Тибе организовали группу, где собираются носители недуга. Как говорит Суда, разговоры помогают облегчить боль, хотя под конец встречи всегда задаётся один и тот же вопрос – «Почему эта болезнь у меня?».
Надежды на лекарство
Человеческие ДНК и гистоны (белки, уплотняющие хромосомы в клетки) могут приобретать химические метки, которые не меняют основу генов, но могут заглушать или усилить активность гена.
На то, где ставятся такие отметки и какие формы они принимают, вероятно влияет поведение и окружение человека. Например, курение или стресс. Возможно, у некоторых отметки появляются случайным образом или в результате мутаций, в том числе при раке. Учёные не знают, как и почему клетки добавляют эти метки, но некоторые из них связаны со старением.
Профессор генетики и биостатистики в Калифорнийском университете Лос-Анджелеса Стив Хорват утверждает, что использовал одну из этих меток для изучения эпигенетических часов – это совокупность меток ДНК, позволяющая определить биологический возраст ткани, клетки или органа. Такие метки можно прочитать даже по образцам крови и мочи. Как показало исследование 18 человек с синдромом Вернера, их клетки имели заметно более высокий эпигенетический возраст, чем у контрольной группы без недуга.
Пониманию тонкостей старения способствует и генетическая информация Нагашимы и Суды, которую они передали в надежде, что это принесёт пользу в будущем.
Учёные уже знают, что ген WRN – это ключ к пониманию работы клеток и ДНК в целом. Известно, что нарушение WRN приводит к широко распространённой нестабильности всего гена.
Главный вопрос заключается в том, являются ли метки на ДНК лишь отпечатками болезней и старения, или же они сами вызывают болезни и старение. Если последнее, то тогда встаёт очевидный вопрос – можно ли редактированием или удалением этих меток предотвратить или обратить вспять часть старения или возрастное заболевание?
В 2006-2007 годы японский исследователь Синъя Яманака (Shinya Yamanaka) впервые в мире получил индуцированные стволовые клетки и опубликовал исследования на тему того, что любую взрослую клетку можно перевести в более ранее эмбриональное состояние, из которого затем её можно превратить в другой тип клетки. За эти достижения Яманака получил Нобелевскую премию, став главной пружиной для исследований стволовых клеток.
За эти достижения Яманака получил Нобелевскую премию, став главной пружиной для исследований стволовых клеток.
Синъя Яманака Фото Reuters
Что особо важно, при исследованиях Яманака смог полностью стереть эпигенетические метки клетки. Позже исследователи повторили это на мышах, которые страдали от синдрома преждевременного старения. В конце опытов мыши ненадолго омолодились, но умерли через несколько дней. Эксперимент также привел к раку и потере у клеток способности функционировать.
В 2016 году учёные из Калифорнийского университета Сан-Диего улучшили методы эксперимента, благодаря чему у подопытных мышей замедлилось преждевременное старение. Животные выглядели более здоровыми и жили на 30% дольше необработанных мышей, также страдавших от синдрома. Более того, у стареющих мышей такое лечение привело к омоложению поджелудочной железы и мышц.
Животные выглядели более здоровыми и жили на 30% дольше необработанных мышей, также страдавших от синдрома. Более того, у стареющих мышей такое лечение привело к омоложению поджелудочной железы и мышц.
Иными словами, опыты идут активно. Но один из главных вопросов заключается в том, связанно ли удаление эпигенетических меток с развитием и старением клетки, или же это просто побочный эффект? Профессор Хорват из Калифорнийского университета считает, что с эпигенетической точки зрения есть явные общие черты старения во многих областях тела. Старение головного мозга сходно со старением печени или почек, потому что в целом процесс старения довольно простой, так как воспроизводим в разных органах.
По словам Хорвата, эта научная область сродни золотой лихорадки – все стремятся её изучить.
 Это не удивительно, ведь когда-то считалось, что человек не может изменить свои гены, но современная наука указывает на другое.
Это не удивительно, ведь когда-то считалось, что человек не может изменить свои гены, но современная наука указывает на другое.Незадолго до диагноза Нагашима собирался жениться, и девушка осталась с ним после приговора врачей. Они даже прошли генетический тест, чтобы убедиться, что не передадут заболевание своим детям. Но когда родители невесты узнали о состоянии японца, они этого не одобрили. На этом отношения закончились.
С тех пор у Нагашимы появилась новая девушка, с которой он хочет связать жизнь. Правда, пока ему не хватает смелости попросить разрешения об этом у родителей возлюбленной. Его тело постоянно болит, а конечности распухают, но с этим он смирился: «Больно. Но я буду терпеть».
Синдром Вернера | Cancer.
 Net
NetЧто такое синдром Вернера?
Синдром Вернера, также называемый прогерией, — это наследственное заболевание, связанное с преждевременным старением и повышенным риском рака и других заболеваний. Признаки синдрома Вернера обычно развиваются в детстве или подростковом возрасте. У человека с синдромом Вернера нет обычного скачка роста, типичного для подростка, и он, как правило, короче, чем другие. Признаки старения, включая морщины, седые волосы и их выпадение, могут появиться у людей в возрасте от 20 лет.В возрасте 30 лет симптомы, которые могут развиться, включают катаракту или помутнение хрусталика глаза, диабет 2 типа, кожные язвы, нос с клювом и остеопороз, что означает снижение минеральной плотности костей. Одной из наиболее серьезных проблем со здоровьем, с которыми сталкиваются люди с синдромом Вернера, является раннее развитие различных типов рака и атеросклероза, обычно известных как уплотнение артерий, которое может привести к сердечному приступу.
Что вызывает синдром Вернера?
Синдром Вернера — это генетическое заболевание. Это означает, что риск синдрома Вернера может передаваться в семье из поколения в поколение. Известно, что мутации (изменения) в гене WRN вызывают синдром Вернера. Исследования продолжаются, чтобы узнать больше о синдроме Вернера.
Это означает, что риск синдрома Вернера может передаваться в семье из поколения в поколение. Известно, что мутации (изменения) в гене WRN вызывают синдром Вернера. Исследования продолжаются, чтобы узнать больше о синдроме Вернера.
Как наследуется синдром Вернера?
Обычно каждая клетка имеет по 2 копии каждого гена: 1 унаследован от матери и 1 унаследован от отца. Синдром Вернера следует аутосомно-рецессивному типу наследования, что означает, что мутация должна присутствовать в обеих копиях гена, чтобы человек был затронут.Это означает, что оба родителя должны передать мутацию гена, чтобы ребенок был затронут. Человека, у которого есть только одна копия мутации гена, называют «носителем». Когда оба родителя являются носителями рецессивной генной мутации, существует 25% -ная вероятность, что ребенок унаследует 2 мутации и пострадает.
Существуют варианты для людей, заинтересованных в рождении ребенка, когда будущий родитель несет генную мутацию, которая увеличивает риск этого наследственного синдрома рака. Преимплантационная генетическая диагностика (ПГД) — это медицинская процедура, проводимая в сочетании с экстракорпоральным оплодотворением (ЭКО).Это позволяет людям, несущим конкретную известную генетическую мутацию, снизить вероятность того, что их дети унаследуют это заболевание. Яйцеклетки женщины удаляются и оплодотворяются в лаборатории. Когда эмбрионы достигают определенного размера, одна клетка удаляется и проверяется на рассматриваемое наследственное заболевание. Затем родители могут выбрать перенос эмбрионов, у которых нет мутации. ПГД используется уже более двух десятилетий и используется для лечения нескольких синдромов наследственной предрасположенности к раку.Однако это сложная процедура с финансовыми, физическими и эмоциональными факторами, которые необходимо учитывать перед началом. Для получения дополнительной информации обратитесь к специалисту по вспомогательной репродукции в клинике репродуктивной медицины.
Насколько распространен синдром Вернера?
Синдром Вернера считается очень редким. По оценкам, 1 из 200 000 человек в США может иметь синдром Вернера. Синдром Вернера несколько чаще встречается в Японии и на Сардинии в Италии, где, по оценкам, это состояние может иметь 1 из 30 000 человек.Это связано с тем, что генетическая мутация, как было известно, происходила много поколений назад, когда их популяция была меньше по численности, и со временем генетическая мутация передавалась неоднократно, затрагивая большее количество людей; это называется мутацией-основателем.
По оценкам, 1 из 200 000 человек в США может иметь синдром Вернера. Синдром Вернера несколько чаще встречается в Японии и на Сардинии в Италии, где, по оценкам, это состояние может иметь 1 из 30 000 человек.Это связано с тем, что генетическая мутация, как было известно, происходила много поколений назад, когда их популяция была меньше по численности, и со временем генетическая мутация передавалась неоднократно, затрагивая большее количество людей; это называется мутацией-основателем.
Как диагностируется синдром Вернера?
В настоящее время диагноз синдрома Вернера подозревается, если кто-то имеет несколько из перечисленных ниже особенностей.
Общие признаки синдрома Вернера (диагностирован после 10 лет):
Катаракта обоих глаз
Изменения кожи, связанные со старением, такие как морщины, истончение, уплотнение, язвы, сухость, изменение цвета, синяки
Характерные черты лица, включая появление морщин и потерю мышечного тонуса
Короткая высота
Раннее поседение или истончение волос
Семейный анамнез синдрома Вернера
Положительный результат суточного теста на гиалуроновую кислоту в моче
Другие признаки синдрома Вернера:
Сахарный диабет 2 типа
Проблемы с фертильностью (возможностью иметь ребенка)
Остеопороз
Раки
Костные изменения пальцев рук и ног
Изменения тканей
Ранний атеросклероз, означающий накопление бляшек в артериях
Хриплый или высокий голос
Плоскостопие и тонкие конечности
Были предложены рекомендации по диагностике синдрома Вернера, но со временем они могут измениться по мере того, как мы узнаем больше об этом состоянии.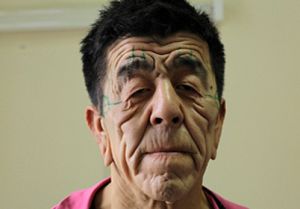 Генетическое тестирование на мутации в гене WRN доступно только в рамках научных исследований (клинических испытаний). Мутации в гене WRN обнаруживаются примерно у 90% людей с синдромом Вернера. Остальные, вероятно, вызваны генными мутациями, которые еще не были обнаружены исследователями.
Генетическое тестирование на мутации в гене WRN доступно только в рамках научных исследований (клинических испытаний). Мутации в гене WRN обнаруживаются примерно у 90% людей с синдромом Вернера. Остальные, вероятно, вызваны генными мутациями, которые еще не были обнаружены исследователями.
Каковы предполагаемые риски рака, связанные с синдромом Вернера?
Риск рака повышен у людей с синдромом Вернера, но конкретный риск рака неизвестен.Типы рака, о которых сообщают у людей с синдромом Вернера, включают рак щитовидной железы, меланому на коже, саркому мягких тканей и остеосаркому (тип рака костей).
Какие есть варианты скрининга синдрома Вернера?
Рекомендуемые обследования для людей с диагнозом синдрома Вернера включают:
Ежегодный скрининг на диабет 2 типа
Годовой липидный профиль по анализу крови
Ежегодное обследование глаз для выявления признаков катаракты
Ежегодный медицинский осмотр с особым вниманием к здоровью сердца и дерматологические (кожные) обследования для выявления рака, связанного с синдромом Вернера
Людям с синдромом Вернера следует также избегать курения и физических травм, быть физически активными и поддерживать здоровый вес, чтобы снизить риск сердечных заболеваний и других возрастных заболеваний. Любые боли в груди, также известные как стенокардия, должны быть тщательно обследованы врачом. Из-за риска рака кожи людям с синдромом Вернера также следует ограничивать пребывание на солнце и защищаться от солнечного света на улице.
Любые боли в груди, также известные как стенокардия, должны быть тщательно обследованы врачом. Из-за риска рака кожи людям с синдромом Вернера также следует ограничивать пребывание на солнце и защищаться от солнечного света на улице.
Рекомендации по скринингу могут меняться со временем по мере развития новых технологий и получения дополнительных сведений о синдроме Вернера. Важно обсудить с врачом соответствующие скрининговые тесты.
Узнайте больше о том, чего ожидать от стандартных тестов, процедур и сканирований.
Вопросы, которые следует задать бригаде здравоохранения
Если вас беспокоит риск заболевания раком, поговорите со своим лечащим врачом. Может быть полезно пригласить кого-нибудь на встречи, чтобы делать записи. Вы можете задать своей медицинской бригаде следующие вопросы:
Каков мой риск рака?
Что я могу сделать, чтобы снизить риск рака?
Какие у меня есть варианты скрининга на рак?
Если вас беспокоит история вашей семьи и вы считаете, что у вас или других членов семьи может быть синдром Вернера, подумайте о том, чтобы задать следующие вопросы:
Увеличивает ли мой семейный анамнез риск рака?
Указывает ли он на необходимость оценки риска рака?
Вы направите меня к генетическому консультанту или другому специалисту по генетике?
Стоит ли рассматривать генетическое тестирование?
Связанные ресурсы
Генетика рака
Генетическое тестирование
Чего ожидать при встрече с консультантом по генетике
Сбор семейной истории рака
Обмен результатами генетического теста с семьей
Вопросы и ответы о семейном генетическом тестировании
Дополнительная информация
Национальная организация редких заболеваний
Чтобы найти консультанта по генетическим вопросам в вашем районе, обратитесь к своей медицинской бригаде или посетите этот веб-сайт:
Национальное общество консультантов по генетике
Понимание фенотипа преждевременного старения — первый случай, описанный в Колумбии
Синдром Вернера (WS) — это аутосомно-рецессивное генетическое заболевание, которое в основном характеризуется изменениями кожи, подобными склеродермии, ювенильной катарактой, низким ростом и признаками преждевременного старения.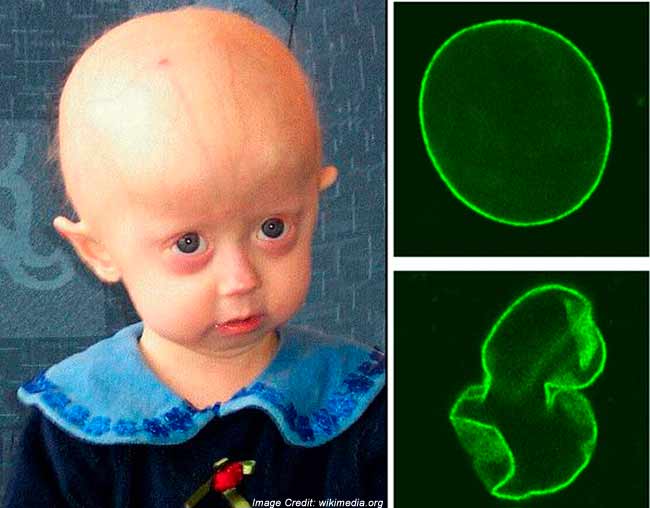 Мы сообщаем о случае 48-летнего пациента мужского пола, у которого имеются основные признаки WS, включая высокий голос, склеротические поражения кожи, в основном на стопах, преждевременное поседение волос на коже головы, двустороннюю катаракту и «птичье» лицо. внешность. Кроме того, у пациента представлены другие клинические характеристики, наблюдаемые у пациентов с WS, такие как низкий рост, сахарный диабет 2 типа, гипогонадизм, родство родителей и наличие в анамнезе братьев или сестер с аналогичными клиническими характеристиками. Секвенирование гена WRN позволило идентифицировать гомозиготный патогенный вариант NM_00553.4: c.2581C> T (NP_000544.2: pGln861Ter). Это первый случай WS, зарегистрированный среди населения Колумбии. Мы сообщаем об этом случае, чтобы избежать неправильного диагноза этого нечастого состояния и позволить своевременно выявить потенциальные осложнения, связанные с преждевременным старением, особенно злокачественные новообразования, сердечно-сосудистые и метаболические заболевания.
Мы сообщаем о случае 48-летнего пациента мужского пола, у которого имеются основные признаки WS, включая высокий голос, склеротические поражения кожи, в основном на стопах, преждевременное поседение волос на коже головы, двустороннюю катаракту и «птичье» лицо. внешность. Кроме того, у пациента представлены другие клинические характеристики, наблюдаемые у пациентов с WS, такие как низкий рост, сахарный диабет 2 типа, гипогонадизм, родство родителей и наличие в анамнезе братьев или сестер с аналогичными клиническими характеристиками. Секвенирование гена WRN позволило идентифицировать гомозиготный патогенный вариант NM_00553.4: c.2581C> T (NP_000544.2: pGln861Ter). Это первый случай WS, зарегистрированный среди населения Колумбии. Мы сообщаем об этом случае, чтобы избежать неправильного диагноза этого нечастого состояния и позволить своевременно выявить потенциальные осложнения, связанные с преждевременным старением, особенно злокачественные новообразования, сердечно-сосудистые и метаболические заболевания.
1. Введение
WS был первоначально описан Отто Вернером в 1904 году. Он сообщил о 4 случаях братьев и сестер с ювенильной катарактой, кожными изменениями конечностей, похожими на склеродермию, деформациями суставов, низким ростом, старческой внешностью, ювенильной седой. и гипоплазия половых органов [1].Со времени первого описания во всем мире были описаны дополнительные сообщения о случаях WS, большинство из которых были из Японии [2, 3]. Распространенность среди населения Японии составляет от 1/20 000 до 1/40 000 [4]. Частота носителей гетерозиготных мутаций оценивается как самая высокая в Японии и Сардинии, составляя 1/166 [4] и 1/120 [5] соответственно. Распространенность среди населения Колумбии неизвестна.
Ген WRN (также называемый RECQL2 или REQ3) в chr 8p12 является единственным известным геном, ответственным за WS. Ген WRN имеет 34 кодирующих экзона, которые кодируют ядерный белок из 1432 аминокислот; этот белок является членом геликаз ДНК RecQ.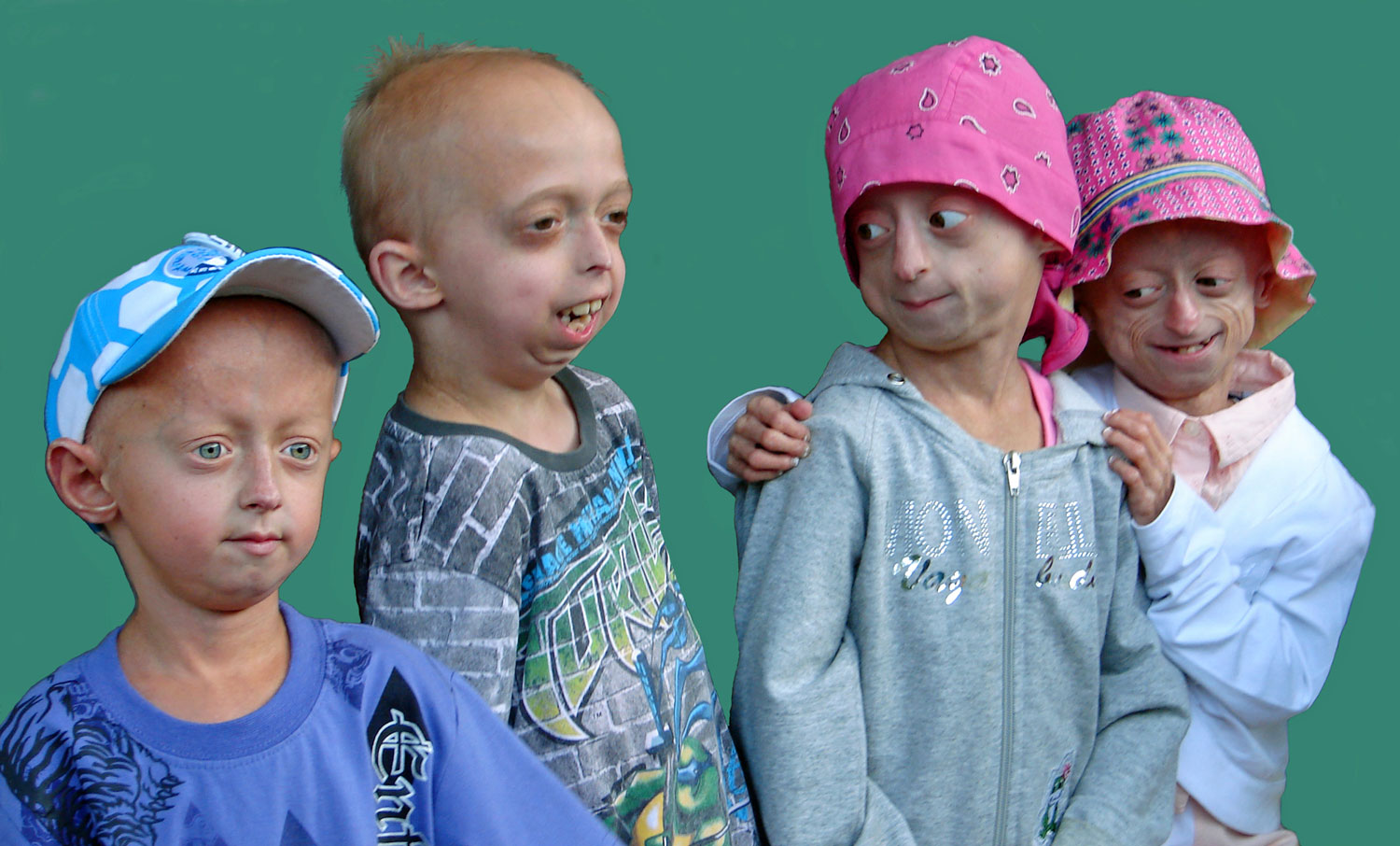 Были проведены многочисленные биохимические и клеточно-биологические исследования для оценки клеточных эффектов, связанных с потерей функции белка WRN. Эти исследования продемонстрировали важность спиральной активности белка WRN для поддержания стабильности генома, включая репарацию ДНК, репликацию, транскрипцию и поддержание теломер [6].
Были проведены многочисленные биохимические и клеточно-биологические исследования для оценки клеточных эффектов, связанных с потерей функции белка WRN. Эти исследования продемонстрировали важность спиральной активности белка WRN для поддержания стабильности генома, включая репарацию ДНК, репликацию, транскрипцию и поддержание теломер [6].
Клетки пациентов с WS были тщательно изучены, и были идентифицированы некоторые аномалии, включая неспособность восстанавливать ДНК с двухцепочечными разрывами, аномальную динамику теломеразы, клетки, испытывающие медленный рост и укороченный жизненный цикл [7].Сообщалось также о других результатах, о нестабильности хромосом, удлинении S-фазы клеточного цикла и нарушениях инициации репликации ДНК [8].
Потеря функции белка WRN вызывает геномную нестабильность, что приводит к накоплению соматических мутаций, неправильному поддержанию теломер, что может привести к клеточной дисфункции, потере пролиферативного гомеостаза или увеличению потери клеток в различных тканях или клеточных линиях. Эти клеточные изменения, вероятно, ответственны за клинические характеристики раннего старения и развития опухолей, наблюдаемые у пациентов с WS [6, 9].
Эти клеточные изменения, вероятно, ответственны за клинические характеристики раннего старения и развития опухолей, наблюдаемые у пациентов с WS [6, 9].
С момента первого описания гена WRN в 1996 г. сообщалось о различных типах мутаций [10]. Гомозиготные или сложные гетерозиготные мутации потери функции в гене WRN вызывают классический WS. В настоящее время зарегистрировано 83 патогенных варианта со всего мира в Международном регистре синдрома Вернера (Сиэтл, Вашингтон) и Японском консорциуме Вернера (Чиба, Япония) [11]. Специфические по этнической принадлежности мутации WRN также были зарегистрированы в некоторых популяциях, включая японцев (ок.3139-1G> C, r.3139 3233del95; c.1105C> T, p.R369; c.3446delA, p.E1149fs), сардинский (c.2089-3024A> G, r.2088 2089ins106), индийский / пакистанский (c.561A> G, r.557-654del98), марокканский (c.2179dupT, p. C727fs), турецкой (c.3460-2A> G, r.3460 3572del113) и голландской (c.3590delA, pN1197fs) популяциями [12, 13].
Большинство патогенных вариантов приводят к усечению белка WRN из-за пропуска экзона, связанного со стоп-кодонами, небольшими вставками / делециями или мутациями сплайсинга. Большинство патогенных вариантов находится в экзонах, но также описаны интронные варианты [12].Эти мутации вызывают потерю сигнала ядерной локализации на C-конце белка WRN и / или способствуют нонсенс-опосредованному распаду мутантной мРНК [11].
Хотя отчет о возможной генотип-фенотипической корреляции фолликулярной карциномы с C-концевой мутацией WRN и папиллярной карциномой с N-концевой мутацией WRN- среди японских пациентов WS был опубликован [14], в целом клинические фенотипы и естественная история пациентов с WS, по-видимому, очень похожи среди типов мутации WRN и различных этнических групп [11].
Пересмотренные диагностические критерии для WS [3] включают следующие основные признаки: прогероидные изменения волос, катаракта, изменения кожи, трудноизлечимые язвы кожи, кальциноз мягких тканей, птичье лицо и ненормальный голос. У этих пациентов могут быть другие ассоциированные признаки и симптомы, такие как нарушение метаболизма глюкозы и / или липидов, деформация и аномалии костей, злокачественные опухоли, родство родителей, преждевременный атеросклероз, гипогонадизм, низкий рост и низкая масса тела.Генетический анализ гена WRN теперь включен в диагностические критерии.
У этих пациентов могут быть другие ассоциированные признаки и симптомы, такие как нарушение метаболизма глюкозы и / или липидов, деформация и аномалии костей, злокачественные опухоли, родство родителей, преждевременный атеросклероз, гипогонадизм, низкий рост и низкая масса тела.Генетический анализ гена WRN теперь включен в диагностические критерии.
У нашего пациента диагноз WS был установлен на основании пересмотренных диагностических критериев WS [3]. У пациента были все основные признаки и симптомы (скудные и седые волосы, двусторонняя катаракта, изменения кожи, трудно поддающиеся лечению кожные язвы, кальциноз ахиллова сухожилия, птичье лицо и высокий голос). Также были обнаружены низкий рост, плоскостопие, туловищное ожирение, сахарный диабет 2 типа, гипертриглицеридемия, гипогонадизм и родство родителей.Подтверждение клинического диагноза было сделано путем анализа гена WRN , который выявил патогенный гомозиготный вариант NM_00553.4: c.2581C> T (NP_000544. 2: pGln861Ter). Эта мутация генерирует стоп-кодон в положении 861 и была классифицирована как патогенная.
2: pGln861Ter). Эта мутация генерирует стоп-кодон в положении 861 и была классифицирована как патогенная.
2. История болезни
Мы представляем случай 48-летнего мужчины, который был обследован службой медицинской генетики, потому что он заметил ослабление своего голоса с высоким тоном с 35 лет, связанное с преждевременным поседением с тех пор. ему 30 лет, а кожные поражения — примерно с 40 лет.В возрасте 32 лет была диагностирована двусторонняя катаракта, а в 44 года ему поставили диагноз сахарный диабет, в настоящее время он принимает пероральные гипогликемические средства. Кроме того, у него есть гипотиреоз и гипертриглицеридемия в лечении и кальцификации ахиллова сухожилия. Пациент одобряет отсутствие скачка роста в раннем подростковом возрасте; тем не менее, окончательный рост такой же, как у остальных трех его братьев и сестер (164 см). Пациент сообщает, что у него не было ребенка по собственному желанию.
Пациент является продуктом союза кровных родителей (троюродных братьев) и имеет 49-летнего брата со схожими клиническими характеристиками, включая изменение голоса с 28 лет, двустороннюю катаракту в 29 лет (впоследствии возникают осложнения из-за язвы роговицы. и в настоящее время официально слеп), и преждевременное поседение с 33 лет, более того, склеродермические изменения кожи с 30 лет и диагноз сахарного диабета 2 типа в 35 лет.Его брат также не поддерживает ребенка по собственному желанию. О других осложнениях, таких как атеросклероз, дислипидемия, гипертония, остеопороз или опухоли, не сообщалось.
и в настоящее время официально слеп), и преждевременное поседение с 33 лет, более того, склеродермические изменения кожи с 30 лет и диагноз сахарного диабета 2 типа в 35 лет.Его брат также не поддерживает ребенка по собственному желанию. О других осложнениях, таких как атеросклероз, дислипидемия, гипертония, остеопороз или опухоли, не сообщалось.
К сожалению, брат и родители пациента отказались от генетического тестирования. Других родственников с клиническим подозрением на WS нет.
Пациент констатирует, что у тети по материнской линии лейкоз неуточненного типа, а у отца в анамнезе — острый инфаркт миокарда в 65 лет и диагноз меланомы в 85 лет. Дяде по матери диагностирован рак легких в 72 года, а у дедушки по материнской линии рак простаты диагностирован в 73 года. .
При первоначальном медицинском осмотре он выглядел намного старше своего возраста с «птичьим» лицом, носом в форме клюва и двусторонней катарактой, его голос был высоким, а его волосы и брови были редкими и заметно седыми. У него были тонкие верхние конечности с пониженным содержанием подкожного жира и туловищным ожирением (рис. 1). Кроме того, мы обнаружили низкий рост, гипогенитализм, нижние конечности с заметно атрофированной кожей и подкожно-жировой клетчаткой, аномальную пигментацию кожи и гиперкератоз, а также плоскостопие (Рисунки 2 и 3).
У него были тонкие верхние конечности с пониженным содержанием подкожного жира и туловищным ожирением (рис. 1). Кроме того, мы обнаружили низкий рост, гипогенитализм, нижние конечности с заметно атрофированной кожей и подкожно-жировой клетчаткой, аномальную пигментацию кожи и гиперкератоз, а также плоскостопие (Рисунки 2 и 3).
Секвенирование гена WRN выявило гомозиготный вариант NM_00553.4: c.2581C> T (NP_000544.2: pGln861Ter). Отчет о секвенировании гена WRN можно найти в дополнительном материале S1. Этот вариант генерирует стоп-кодон в положении 861 и был классифицирован как патогенный и ранее описанный в гомозиготном статусе у пациента европеоидной расы из США в 2006 году [15].
2.1. Исследования
Лабораторные данные включали нормальную функцию почек, высокий уровень глюкозы в крови (164 мг / дл), повышенный уровень гликозилированного гемоглобина (9.4%) и повышенных триглицеридов (324,6 мг / дл) с нормальным холестерином (162,4 мг / дл). ЭКГ показала подъем точки J за счет ранней реполяризации. КТ брюшной полости показала двусторонние кисты почек, небольшую пупочную грыжу и отсутствие жировой ткани в печени. УЗИ яичек показало уменьшение двустороннего объема яичек, в основном с левой стороны.
ЭКГ показала подъем точки J за счет ранней реполяризации. КТ брюшной полости показала двусторонние кисты почек, небольшую пупочную грыжу и отсутствие жировой ткани в печени. УЗИ яичек показало уменьшение двустороннего объема яичек, в основном с левой стороны.
2.2. Результат и наблюдение
Пациентам с WS рекомендуется регулярный скрининг на злокачественные новообразования из-за высокого риска ранних новообразований.Также очень важно исключить сердечно-сосудистые и метаболические заболевания во время наблюдения за этими пациентами. Наш пациент все еще находится под периодическим клиническим наблюдением и наблюдением. В настоящее время он проходит лечение пероральными гипогликемическими средствами от СД2 с адекватным контролем уровня глюкозы и лечит гипертриглицеридемию. До сих пор не обнаружено никаких признаков атеросклероза или сердечно-сосудистых заболеваний. Однако недавно ему поставили диагноз «рефрактерная цитопения с многолинейной дисплазией», формой миелодиспластического синдрома, которая потребовала нескольких переливаний.
Согласно истории болезни, брат пациента находится под наблюдением на предмет недостаточного контроля сахарного диабета и тяжелых поражений кожи, которые трудно лечить, но рак не зарегистрирован.
3. Обсуждение
Первым клиническим признаком WS, часто распознаваемым ретроспективно, является отсутствие ожидаемого скачка пубертатного роста, ведущего к относительно низкому росту в зрелом возрасте. Однако иногда этот клинический признак упускается из виду, как правило, в раннем взрослом возрасте (36.7_ ± 10,1 года) [2] при постановке диагноза в связи с другими классическими признаками. Пациенты с WS являются нормальными при рождении и имеют адекватный рост и развитие в детстве. После этого у пациентов постепенно развиваются типичные черты WS, такие как пожилой внешний вид, который включает птичье лицо, седые волосы, алопецию, атрофию кожи и потерю подкожного жира, а также области гипо- и гиперпигментации.
Осложнения, обычно начинающиеся в возрасте 30 лет, такие как двусторонняя катаракта, артериосклеротические заболевания (церебральное кровоизлияние, инфаркт мозга, инфаркт миокарда и облитерирующий артериосклероз), гипертония, сахарный диабет, дислипидемия, остеопороз, глубокая язва кожи вокруг лодыжки. Ахиллово сухожилие, злокачественные новообразования и ранняя потеря бесплодия, связанная с атрофией гонад.Основными причинами смерти в среднем возрасте 54 лет являются инфаркт миокарда, вторичный по отношению к атеросклерозу, сахарный диабет и злокачественные опухоли [6].
Ахиллово сухожилие, злокачественные новообразования и ранняя потеря бесплодия, связанная с атрофией гонад.Основными причинами смерти в среднем возрасте 54 лет являются инфаркт миокарда, вторичный по отношению к атеросклерозу, сахарный диабет и злокачественные опухоли [6].
Пациенты WS имеют гораздо более высокую частоту новообразований, а средний возраст при первом диагнозе новообразования составляет 43,3 ± 9,9 года (диапазон 20–69), что подтверждается систематическим обзором литературы, проведенным Lauper et al. [16]. Основными признаками рака при WS являются ранний возраст начала, высокая частота необычных типов, особенно сарком, и множественных новообразований [17]; однако описаны и распространенные типы рака.
В исследовании Лаупера [16] было проанализировано 189 пациентов с WS с 248 новообразованиями, чтобы охарактеризовать спектр неоплазии в WS; 139 (74%) из них были резидентами Японии. Они обнаружили, что множественные новообразования наблюдались у 22% пациентов с WS, а наиболее частыми новообразованиями были новообразования щитовидной железы (16,1%), за которыми следовали злокачественная меланома (13,3%), менингиома (10,9%), саркомы мягких тканей (10,1%), лейкоз и связанные с ним гематологические нарушения (9,3%) и остеосаркома (7,7%). Риск рака был значительно повышен у проживающих в Японии пациентов WS для шести наиболее частых новообразований, за исключением лейкемии, а повышенный риск этих новообразований колеблется от 8.9 для новообразований щитовидной железы и в 53 раза выше для меланом, чем в контрольной популяции.
Риск рака был значительно повышен у проживающих в Японии пациентов WS для шести наиболее частых новообразований, за исключением лейкемии, а повышенный риск этих новообразований колеблется от 8.9 для новообразований щитовидной железы и в 53 раза выше для меланом, чем в контрольной популяции.
Мы хотели бы внести свой вклад в литературу нашим клиническим наблюдением за классическим случаем WS; этому пациенту был поставлен диагноз относительно поздно, потому что изначально этот синдром не предполагался, возможно, из-за плохой осведомленности об этом редком заболевании, которое приводит к симптоматическому лечению каждого проявления. Хотя первым клиническим признаком WS, часто распознаваемым ретроспективно, является отсутствие ожидаемого всплеска пубертатного роста, типичные признаки WS постепенно появляются после полового созревания.Поэтому у молодых пациентов некоторые симптомы могут отсутствовать, и это может отсрочить постановку диагноза. Это демонстрирует, что знание ранних признаков WS и семейного анамнеза может быть полезным для раннего распознавания WS и установления диагноза.
Это первый зарегистрированный случай синдрома Вернера в колумбийской популяции, у которой клинический фенотип аналогичен ранее зарегистрированным в других популяциях. Мы сообщаем об этом случае, чтобы избежать неправильного диагноза этого нечастого состояния и позволить своевременно выявить потенциальные осложнения, связанные с преждевременным старением, особенно злокачественные новообразования, сердечно-сосудистые и метаболические заболевания.
Очки обучения (i) WS следует подозревать при наличии кардинальных признаков, таких как изменения голоса, склеродермические изменения кожи, двусторонняя катаракта, кальцификация мягких тканей и появление преждевременного старения. (Ii) Это важно. распознать это заболевание на ранней стадии с целью скрининга и выявления злокачественных опухолей и других осложнений, таких как сердечно-сосудистые заболевания, которые обычно связаны с возрастом и могут потенциально угрожать жизни. (iii) Многопрофильное ведение таких пациентов имеет важное значение для лечения и профилактика сопутствующих осложнений. (iv) Прогноз определяется тяжестью осложнений, связанных с синдромом, таких как инфаркт миокарда, инсулинорезистентность и риск рака. (v) Пациентам с WS рекомендуется контролировать годовой уровень глюкозы и липидов, офтальмологическое обследование и полное физикальное обследование для выявления возможных ранних проявлений наиболее частых осложнений WS.
Конфликт интересов
Эта работа не поддерживается грантом. Авторы заявляют об отсутствии конфликта интересов при подготовке рукописи.
Дополнительные материалы
S1 : Отчет о секвенировании гена WRN . Описание методологии и интерпретация обнаруженного патогенного варианта. (Дополнительные материалы)
Синдром Вернера: общие сведения, патофизиология, эпидемиология
Автор
Камила К. Джаннигер, доктор медицины клинический профессор дерматологии, клинический доцент педиатрии, заведующий педиатрической дерматологией, Медицинская школа Рутгерса, Нью-Джерси
Камила К. Джаннигер, доктор медицины, является членом следующих медицинских обществ: Американская академия дерматологии
Джаннигер, доктор медицины, является членом следующих медицинских обществ: Американская академия дерматологии
Раскрытие: Ничего не раскрывать.
Соавтор (ы)
Анна Возняцка, MD, PhD Профессор кафедры дерматологии, Медицинский университет Лодзи, Польша
Анна Возняцка, MD, PhD является членом следующих медицинских обществ: Европейская академия дерматологии и венерологии, Польское дерматологическое общество
Раскрытие: нечего раскрывать.
Специальная редакционная коллегия
Майкл Дж. Уэллс, доктор медицины, FAAD Дерматологический хирург / хирург Мооса, Хирургический центр в Plano Dermatology
Майкл Дж. Уэллс, доктор медицины, FAAD является членом следующих медицинских обществ: Alpha Omega Alpha, Американская академия дерматологии, American Medical Ассоциация, Техасская медицинская ассоциация
Раскрытие информации: не подлежит разглашению.
Роберт А. Шварц, доктор медицины, магистр здравоохранения Профессор и руководитель дерматологии, профессор патологии, профессор педиатрии, профессор медицины, Медицинская школа Рутгерса, Нью-Джерси
Роберт А. Шварц, доктор медицины, магистр здравоохранения является членом следующих медицинских обществ : Alpha Omega Alpha, Американская академия дерматологии, Нью-Йоркская медицинская академия, Королевский колледж врачей Эдинбурга, Sigma Xi, The Scientific Research Honor Society
Раскрытие: Ничего не раскрывать.
Главный редактор
Дирк М. Элстон, доктор медицины Профессор и заведующий кафедрой дерматологии и дерматологической хирургии Медицинского колледжа Медицинского университета Южной Каролины
Дирк Элстон, доктор медицины, является членом следующих медицинских обществ: Американская академия дерматологии
Раскрытие информации : Нечего раскрывать.
Дополнительные участники
Франклин Флауэрс, доктор медицины Почетный профессор кафедры дерматологии, аффилированный доцент кафедры патологии Медицинского колледжа Университета Флориды
Франклин Флауэрс, доктор медицины, является членом следующих медицинских обществ: Американский колледж хирургии Мооса
Раскрытие информации: Нечего раскрывать.
| Трихотиодистрофия (TTD) | TFIIH , XPB , XPD | TC-NER, транскрипция | Неврологический и скелетно-диастрофический дерматоз бесплодие и ломкие волосы и ногти | [46] |
| Синдром Кокейна (CS) | CSA или CSB | TC-NER | Кахексия, дегенерация нейронов, потеря клеток сетчатки, замедленный рост катаракта, светочувствительность, атеросклероз, диабет, гипертония | [46] |
| Xeroderma pigmentosum (XP) | XPA-XPG | NER | Симптомы CS и, кроме того: гиперчувствительность к воздействию УФ-излучения высокая частота рака кожи | [46] |
| Атаксия телеангиэктазия (AT) | ATM | DDR | Прогрессирующая дегенерация мозжечка, тяжелая атаксия, задержка роста, расширение кровеносных сосудов, иммунологические дефекты и рак | [51] |
| Синдром Ротмунда – Томсона (RTS) | RECQL4 | Дефицит роста ДНК | , седые волосы, катаракта, пойкилодермия, остеосаркомы и рак кожи[28] | |
| Синдром Вернера (WS) | WRN | Поддержание теломер, рекомбинация и восстановление ДНК | Атрофическая кожа, атрофическая кожа остеопороз, диабет II типа, аутоиммунитет, атрофия кожи и мышц, плохое заживление ран, катаракта, атеросклероз, гипогонадизм и рак | [27,28] |
| Синдром Блума (BS) | BLM | Митотическая рекомбинация | Задержка роста, чувствительность к солнцу, иммунодефицит, геномная нестабильность, рак и диабет | [28] |
| Синдром прогерии Хатчинсона-Гилфорда (HGPS) | LMNA | Функция ядерной пластинки | Алопеция, саркопения, атеросклероз, остеолиз, выступающие вены черепа, потеря подкожного жира, проблемы с высоким уровнем полового развития и сосудистые проблемы голос | [59] |
| Врожденный дискератоз (DC) | DKC1 | Поддержание теломер | Задержка роста, микроцефалия, гипоплазия мозжечка, умственная отсталость3, прогрессирующая комбинированная недостаточность иммунитета | [].

Синдром Вернера — обзор
Синдром Вернера
Синдром Вернера, или прогерия у взрослых, является аутосомно-рецессивным заболеванием, которое связано с мутациями гена WRN , расположенного на хромосоме 8p11-p12. 95 Ген WRN имеет решающее значение для репликации и восстановления ДНК. Мутации в гене WRN приводят к усеченным нефункциональным белкам WRN, что в конечном итоге приводит к снижению пролиферации клеток.
Синдром Вернера характеризуется преждевременным старением, которое начинается в третьем десятилетии, со средней продолжительностью жизни 54 года. 70,96,97 Клинические проявления включают тонкую кожу, морщины, алопецию, мышечную атрофию, низкий рост из-за отсутствия периода полового созревания, возрастные расстройства, такие как диабет, остеопороз, катаракта и заболевания периферических сосудов, а также различные заболевания. злокачественные опухоли (Таблица 4.1). 96,97
Пациенты с синдромом Вернера имеют повышенный риск как доброкачественных, так и злокачественных заболеваний щитовидной железы. 48 В исследовании японских пациентов с синдромом Вернера возраст диагноза DTC был на 10 лет раньше, и у него было непропорционально большое количество больных мужчин по сравнению с населением Японии в целом. 98 PTC был связан с N-концевым вариантом в WNT , тогда как FTC чаще наблюдается с C-концевым вариантом. 98 Эти пациенты имеют необычное распределение гистологических подтипов DTC с более высокими показателями FTC (48% при синдроме Вернера против 14% среди населения Японии в целом) и анапластического рака щитовидной железы (13% против 2% соответственно). и более низкий уровень классического PTC (35% против 78% соответственно). 98 В целом риск развития NMTC для пациентов с синдромом Вернера оценивается в 8,9–16,1%. 99
48 В исследовании японских пациентов с синдромом Вернера возраст диагноза DTC был на 10 лет раньше, и у него было непропорционально большое количество больных мужчин по сравнению с населением Японии в целом. 98 PTC был связан с N-концевым вариантом в WNT , тогда как FTC чаще наблюдается с C-концевым вариантом. 98 Эти пациенты имеют необычное распределение гистологических подтипов DTC с более высокими показателями FTC (48% при синдроме Вернера против 14% среди населения Японии в целом) и анапластического рака щитовидной железы (13% против 2% соответственно). и более низкий уровень классического PTC (35% против 78% соответственно). 98 В целом риск развития NMTC для пациентов с синдромом Вернера оценивается в 8,9–16,1%. 99
Учитывая распространенность NMTC при синдроме Вернера и высокий уровень агрессивных подтипов, таких как FTC и ATC, скрининг и наблюдение за патологией щитовидной железы в этой популяции оправданы. 100 Диагностическая оценка и лечение заболеваний щитовидной железы, связанных с синдромом Вернера, должны соответствовать стандартным руководящим принципам ведения.
100 Диагностическая оценка и лечение заболеваний щитовидной железы, связанных с синдромом Вернера, должны соответствовать стандартным руководящим принципам ведения.
Хотя для большинства синдромных FNMTC, по-видимому, существует повышенный риск NMTC, пенетрантность низкая и имеется большая фенотипическая гетерогенность. Имеются ограниченные данные о роли скрининга и эпиднадзора, а также о естественном течении NMTC при синдромальном FNMTC. Благодаря будущим исследованиям, направленным на решение этих критических вопросов, включая обнаружение любой ассоциации генотип-фенотип, возможно, удастся усовершенствовать лечение синдромальной FNMTC и оптимизировать исход для пациентов.
Запись OMIM — # 277700
Бэрд, Д. М., Дэвис, Т., Роусон, Дж., Джонс, К. Дж., Киплинг, Д. Нормальная скорость эрозии теломер на уровне отдельных клеток в клетках фибробластов синдрома Вернера. Гм. Molec. Genet. 13: 1515-1524, 2004.
[PubMed: 15150162]
[Полный текст: https: // академ.oup.com/hmg/article-lookup/doi/10.1093/hmg/ddh259]
Гм. Molec. Genet. 13: 1515-1524, 2004.
[PubMed: 15150162]
[Полный текст: https: // академ.oup.com/hmg/article-lookup/doi/10.1093/hmg/ddh259]
Бауэр, Э.А., Сильверман, Н., Бусик, Д.Ф., Кронбергер, А., Деуэл, Т.Ф. Пониженный ответ фибробластов синдрома Вернера на факторы роста PDGF и FGF. Наука 234: 1240-1243, 1986.
[PubMed: 3022382]
[Полный текст: https: // www.sciencemag.org/cgi/pmidlookup?view=long&pmid=3022382]
Наука 234: 1240-1243, 1986.
[PubMed: 3022382]
[Полный текст: https: // www.sciencemag.org/cgi/pmidlookup?view=long&pmid=3022382]
Бойд, М. В. Дж., Грант, А. П. Синдром Вернера (прогерия взрослого): дальнейшие патологические и биохимические наблюдения. Брит. Med. J. 2: 920-925, 1959.
[PubMed: 13803556]
[Полный текст: https://dx.doi.org/10.1136/bmj.2.5157.920]
Брит. Med. J. 2: 920-925, 1959.
[PubMed: 13803556]
[Полный текст: https://dx.doi.org/10.1136/bmj.2.5157.920]
Черимеле, Д., Cottoni, F., Scappaticci, S., Rabbiosi, G., Borroni, G., Sanna, E., Zei, G., Fraccaro, M. Высокая распространенность синдрома Вернера на Сардинии: описание шести пациентов и оценка частоты гена. Гм. Genet. 62: 25-30, 1982.
[PubMed: 7152524]
[Полный текст: https: // dx.doi.org/10.1007/BF00295600]
Гм. Genet. 62: 25-30, 1982.
[PubMed: 7152524]
[Полный текст: https: // dx.doi.org/10.1007/BF00295600]
Чанг, М., Бурмер, Г. К., Суизи, Дж., Лоеб, Л. А., Эдельхофф, С., Дистеч, К. М., Ю, К.-Э., Андерсон, Л., Осима, Дж., Накура, Дж., Мики, Т., Камино, К. , Огихара, Т., Шелленберг, Г. Д., Мартин, Г. М. Доказательства против ДНК-полимеразы бета в качестве гена-кандидата для синдрома Вернера. Гм. Genet. 93: 507-512, 1994.
[PubMed: 8168825]
[Полный текст: https: // dx.doi.org/10.1007/BF00202813]
, Огихара, Т., Шелленберг, Г. Д., Мартин, Г. М. Доказательства против ДНК-полимеразы бета в качестве гена-кандидата для синдрома Вернера. Гм. Genet. 93: 507-512, 1994.
[PubMed: 8168825]
[Полный текст: https: // dx.doi.org/10.1007/BF00202813]
Краббе, Л. , Эмили, А., Нэгер, К. М., Хольтгрев-Грез, Х., Карлседер, Дж. Дисфункция теломер как причина геномной нестабильности при синдроме Вернера. Proc. Nat. Акад. Sci. 104: 2205-2210, 2007.
[PubMed: 17284601]
[Полный текст: http://www.pnas.org/lookup/pmidlookup?view=long&pmid=17284601]
, Эмили, А., Нэгер, К. М., Хольтгрев-Грез, Х., Карлседер, Дж. Дисфункция теломер как причина геномной нестабильности при синдроме Вернера. Proc. Nat. Акад. Sci. 104: 2205-2210, 2007.
[PubMed: 17284601]
[Полный текст: http://www.pnas.org/lookup/pmidlookup?view=long&pmid=17284601]
Эпштейн, К. Дж., Мартин, Г. М., Шульц, А. Л., Мотульский, А. Г. Синдром Вернера: обзор его симптоматики, естественного течения, патологических особенностей, генетики и связи с естественным процессом старения. Медицина 45: 177-222, 1966.
[PubMed: 5327241]
[Полный текст: https: // dx.doi.org/10.1097/00005792-196605000-00001]
Дж., Мартин, Г. М., Шульц, А. Л., Мотульский, А. Г. Синдром Вернера: обзор его симптоматики, естественного течения, патологических особенностей, генетики и связи с естественным процессом старения. Медицина 45: 177-222, 1966.
[PubMed: 5327241]
[Полный текст: https: // dx.doi.org/10.1097/00005792-196605000-00001]
Фарагер, Р. Г. А., Килл, И. Р., Хантер, Дж. А. А., Поуп, Ф. М., Тэннок, К., Должен, С. Ген, ответственный за синдром Вернера, может быть «счетным» геном клеточного деления. Proc. Nat. Акад. Sci. 90: 12030-12034, 1993.
[PubMed: 8265666]
[Полный текст: http: // www.pnas.org/cgi/pmidlookup?view=long&pmid=8265666]
Г. А., Килл, И. Р., Хантер, Дж. А. А., Поуп, Ф. М., Тэннок, К., Должен, С. Ген, ответственный за синдром Вернера, может быть «счетным» геном клеточного деления. Proc. Nat. Акад. Sci. 90: 12030-12034, 1993.
[PubMed: 8265666]
[Полный текст: http: // www.pnas.org/cgi/pmidlookup?view=long&pmid=8265666]
Фукути, К. , Мартин, Г. М., Моннат, Р. Дж., Мл. Мутаторный фенотип синдрома Вернера характеризуется обширными делециями. Proc. Nat. Акад. Sci. 86: 5893-5897, 1989. Примечание: Erratum: Proc. Nat. Акад. Sci. 86: 7994 только, 1989.
[PubMed: 2762303]
[Полный текст: http: // www.pnas.org/cgi/pmidlookup?view=long&pmid=2762303]
Фукути, К. , Танака, К., Кумахара, Ю., Марумо, К., Прайд, М. Б., Мартин, Г. М., Моннат, Р. Дж., Мл. Повышенная частота резистентных к 6-тиогуанину лимфоцитов периферической крови у пациентов с синдромом Вернера. Гм. Genet. 84: 249-252, 1990.
[PubMed: 2303247]
[Полный текст: https: // dx.doi.org/10.1007/BF00200569]
, Танака, К., Кумахара, Ю., Марумо, К., Прайд, М. Б., Мартин, Г. М., Моннат, Р. Дж., Мл. Повышенная частота резистентных к 6-тиогуанину лимфоцитов периферической крови у пациентов с синдромом Вернера. Гм. Genet. 84: 249-252, 1990.
[PubMed: 2303247]
[Полный текст: https: // dx.doi.org/10.1007/BF00200569]
Гебхарт, Э.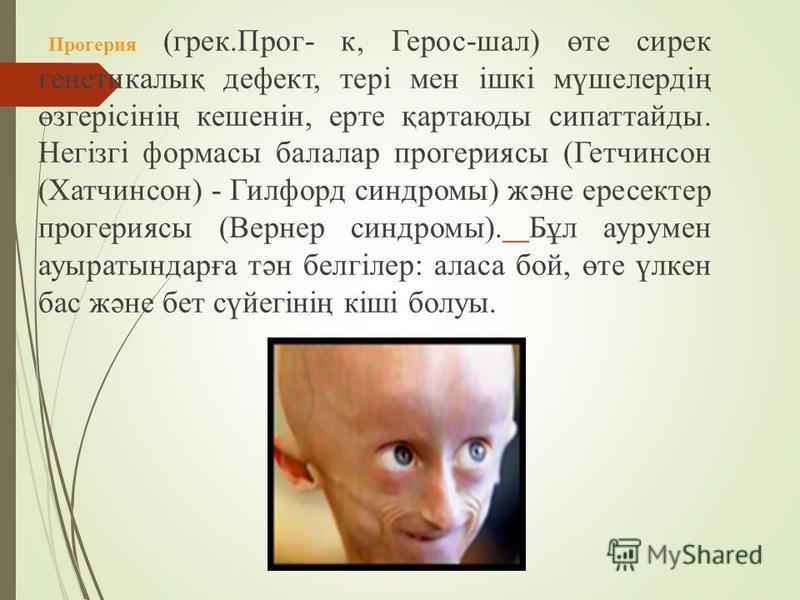 , Бауэр, Р., Рауб, У., Шинцель, М., Рупрехт, К. В., Йонас, Дж. Б. Спонтанная и индуцированная хромосомная нестабильность при синдроме Вернера. Гм. Genet. 80: 135-139, 1988.
[PubMed: 2459043]
[Полный текст: https://dx.doi.org/10.1007/BF00702855]
, Бауэр, Р., Рауб, У., Шинцель, М., Рупрехт, К. В., Йонас, Дж. Б. Спонтанная и индуцированная хромосомная нестабильность при синдроме Вернера. Гм. Genet. 80: 135-139, 1988.
[PubMed: 2459043]
[Полный текст: https://dx.doi.org/10.1007/BF00702855]
Гебхарт, Э.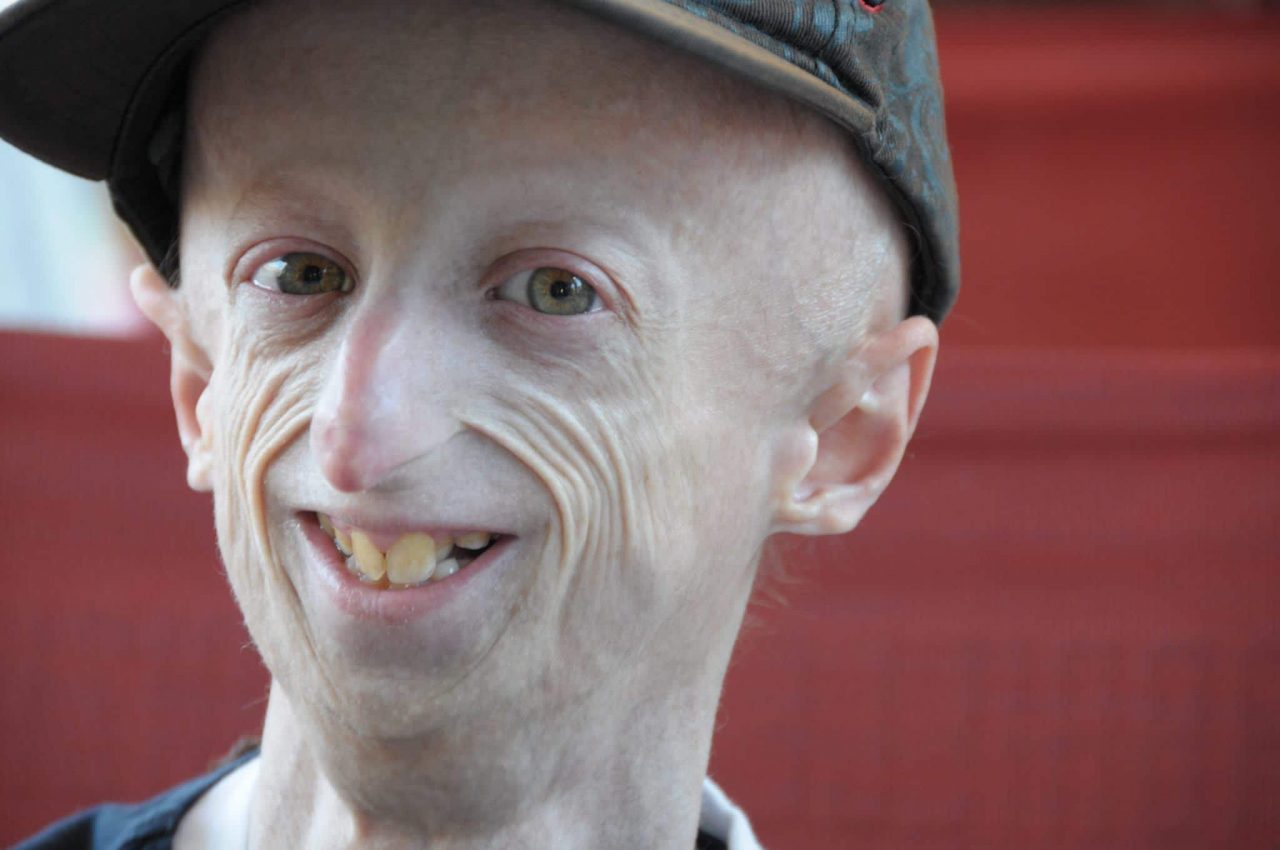 , Шинцель, М., Рупрехт, К. В. Цитогенетические исследования с использованием различных кластогенов у двух пациентов с синдромом Вернера и контрольных лиц. Гм. Genet. 70: 324-327, 1985.
[PubMed: 4018799]
[Полный текст: https: // dx.doi.org/10.1007/BF00295370]
, Шинцель, М., Рупрехт, К. В. Цитогенетические исследования с использованием различных кластогенов у двух пациентов с синдромом Вернера и контрольных лиц. Гм. Genet. 70: 324-327, 1985.
[PubMed: 4018799]
[Полный текст: https: // dx.doi.org/10.1007/BF00295370]
Гото, М., Хариучи, Ю., Танимото, К., Исии, Т., Накашима, Х. Синдром Вернера: разбор 15 случаев с обзором японской литературы. Варенье. Гериат. Soc. 26: 341-347, 1978. [PubMed: 670621] [Полный текст: https://onlinelibrary.wiley.com/resolve/openurl?genre=article&sid=nlm:pubmed&issn=0002-8614&date=1978&volume=26&issue=8&spage=341]
Гото, М., Имамура, О., Куромицу, Дж., Мацумото, Т., Ямабе, Ю., Токутакэ, Ю., Судзуки, Н., Мейсон, Б., Драйна, Д., Сугавара, М., Сугимото, М. , Фуруичи Ю. Анализ мутаций гена геликазы у пациентов с японским синдромом Вернера. Гм. Genet. 99: 191-193, 1997. [PubMed:
336]
Гото М., Миллер Р. В., Исикава Ю., Сугано Х. Избыток редких видов рака при синдроме Вернера (прогерия у взрослых). Cancer Epidemiol. Биомаркеры Пред. 5: 239-246, 1996. [PubMed: 8722214] [Полный текст: http://cebp.aacrjournals.org/cgi/pmidlookup?view=long&pmid=8722214]
Гото, М., Рубинштейн, М., Вебер, Дж., Вудс, К., Драйна, Д. Генетическая связь синдрома Вернера с пятью маркерами на хромосоме 8. Nature 355: 735-738, 1992. [PubMed: 1741060] [Полный текст: https: // doi.org / 10.1038 / 355735a0]
Гото, М., Танимото, К., Хориучи, Ю., Сасадзуки, Т. Семейный анализ синдрома Вернера: опрос 42 японских семей с обзором литературы. Clin. Genet. 19: 8-15, 1981. [PubMed: 7460386] [Полный текст: https://onlinelibrary.wiley.com/resolve/openurl?genre=article&sid=nlm:pubmed&issn=0009-9163&date=1981&volume=19&issue=1&spage=8]
Грей, М.Д., Шен, Ж.-К., Камат-Лоеб, А.С., Бланк, А., Софер, Б.Л., Мартин, Г.М., Осима, Дж., Лоеб, Л.А. Белок синдрома Вернера — это ДНК-геликаза. Nature Genet. 17: 100-103, 1997. [PubMed: 9288107] [Полный текст: https: // doi.org / 10.1038 / ng0997-100]
Хейфлик, Л. Ограниченное время жизни штаммов диплоидных клеток человека in vitro. Exp. Cell Res. 37: 614-636, 1965. [PubMed: 14315085] [Полный текст: https://dx.doi.org/10.1016/0014-4827(65)-9]
Хон, Х., Брайант, Э. М., Ау, К., Норвуд, Т. Х., Боман, Х., Мартин, Г. М. Пестрый транслокационный мозаицизм в культурах фибробластов кожи человека. Cytogenet. Cell Genet. 15: 282-298, 1975. [PubMed: 1222585] [Полный текст: https: // dx.doi.org/10.1159/000130526]
Хуанг, С., Ли, Б., Грей, М. Д., Осима, Дж., Миан, И. С., Кампизи, Дж. Белок синдрома преждевременного старения, WRN, представляет собой экзонуклеазу с 3 примерами и 5 примерами. Nature Genet. 20: 114-116, 1998. [PubMed: 9771700] [Полный текст: https://doi.org/10.1038/2410]
Имамура, О., Итикава, К., Ямабе, Ю., Гото, М., Сугавара, М., Фуруичи, Ю. Клонирование мышиного гомолога гена синдрома Вернера человека и отнесение к 8A4 путем флуоресцентной гибридизации in situ. Геномика 41: 298-300, 1997. [PubMed:
Исикава, Ю., Сугано, Х., Мацумото, Т., Фуруичи, Ю., Миллер, Р. В., Гото, М. Необычные признаки карциномы щитовидной железы у японских пациентов с синдромом Вернера и возможные связи генотип-фенотип с типом клеток и расой. Рак 85: 1345-1352, 1999. [PubMed: 10189141]
Изумино, К., Сакамаки, Х., Исибаши, М., Такино, Х., Ямасаки, Х., Ямагути, Ю., Чикуба, Н., Мацумото, К., Акадзава, С., Токуяма, К., Нагатаки, С. Троглитазон улучшает инсулинорезистентность у пациентов с синдромом Вернера. J. Clin. Endocr. Метаб. 82: 2391-2395, 1997. [PubMed: 9253306] [Полный текст: https: // академ.oup.com/jcem/article-lookup/doi/10.1210/jcem.82.8.4162]
Дженсен, Р. Х., Бигби, В. Л. Прямое иммунофлуоресцентное мечение обеспечивает улучшенный метод анализа соматических мутаций гликофорина А. Cytometry 23: 337-343, 1996. [PubMed: 8
7] [Полный текст: https://doi.org/10.1002/(SICI)1097-0320(19960401)23:4<337::AID-CYTO10>3.0.CO; 2-U]Храйши, М., Ховард, Б., Литтл, Х. Пациент с синдромом Вернера и остеосаркомой, проявляющейся склеродермией. J. Rheum. 19: 810-813, 1992. [PubMed: 1613716]
Крейчи, Л., Ван Комен, С., Ли, Ю., Виллемейн, Дж., Редди, М.С., Кляйн, Х., Элленбергер, Т., Сунг, П. ДНК-геликаза Srs2 разрушает пресинаптический филамент Rad51. Nature 423: 305-309, 2003. [PubMed: 12748644] [Полный текст: https: // doi.org / 10.1038 / nature01577]
Кинг, К. Дж., Мэй, А., Колвраа, С., Бор, В. А. Профили экспрессии генов при синдроме Вернера очень похожи на профили нормального старения. Proc. Nat. Акад. Sci. 100: 12259-12264, 2003. [PubMed: 14527998] [Полный текст: http://www.pnas.org/cgi/pmidlookup?view=long&pmid=14527998]
Ломбард, Д.Б., Берд, К., Джонсон, Б., Марчиньяк, Р.А., Даусман, Дж., Бронсон, Р., Бульманн, Дж. Э., Липман, Р., Карри, Р., Шарп, А., Яениш, Р. , Гуаренте, Л. Мутации в гене WRN у мышей ускоряют смертность на фоне нулевого р53. Molec. Cell. Биол. 20: 3286-3291, 2000. [PubMed: 10757812] [Полный текст: http: // mcb.asm.org/cgi/pmidlookup?view=long&pmid=10757812]
Мартин, Г. М. Мутация Вернера: приводит ли она к «общественному» или «частному» механизму старения? Molec.Med. 3: 356-358, 1997. [PubMed: 9234240]
Мацумото, Т., Симамото, А., Гото, М., Фуруичи, Ю. Нарушение ядерной локализации дефектных ДНК-геликаз при синдроме Вернера. (Письмо) Nature Genet. 16: 335-336, 1997. [PubMed: 9241267] [Полный текст: https: // doi.org / 10.1038 / ng0897-335]
МакКусик, В.А. Медицинская генетика 1962. J. Chronic Dis. 16: 457-634, 1963. [PubMed: 14042963] [Полный текст: https://dx.doi.org/10.1016/0021-9681(63)
Мейслитцер, К., Руппич, В., Вейрих-Швайгер, Х., Вейрих, Х.Г., Ябковский, Дж., Кляйн, Г., Швайгер, М., Хирш-Кауфманн, М. Синдром Вернера: характеристика мутаций в гене WRN в пострадавшей семье. Europ. J. Hum. Genet. 5: 364-370, 1997. [PubMed: 9450180]
Мохагех, П., Хиксон, И.Д. Дефицит ДНК-геликазы, связанный с предрасположенностью к раку и нарушениями преждевременного старения. Гм. Molec. Genet. 10: 741-746, 2001. [PubMed: 11257107] [Полный текст: https: // академ.oup.com/hmg/article-lookup/doi/10.1093/hmg/10.7.741]
Моннат, Р. Дж., Младший, Хакманн, А. Ф. М., Кьяверотти, Т. А. Анализ нуклеотидной последовательности делеций гена гипоксантинфосфорибозилтрансферазы (HPRT) человека. Геномика 13: 777-787, 1992. [PubMed: 1639404] [Полный текст: https://linkinghub.elsevier.com/retrieve/pii/0888-7543(92)-J]
Монна, Р.J., Jr. Необычные признаки карциномы щитовидной железы у японских пациентов с синдромом Вернера и возможные связи генотип-фенотип с типом клеток и расой. (Письмо) Рак 86: 728-729, 1999. [PubMed: 10440702]
Мозер, М.Дж., Бигби, У. Л., Грант, С. Г., Эмонд, М. Дж., Ланглуа, Р. Г., Дженсен, Р. Х., Осима, Дж., Моннат, Р. Дж., Мл. Генетическая нестабильность и риск гематологических заболеваний у пациентов с синдромом Вернера и гетерозигот. Cancer Res. 60: 2492-2496, 2000. [PubMed: 10811130] [Полный текст: http: // Canceres.aacrjournals.org/cgi/pmidlookup?view=long&pmid=10811130]
Мозер, М. Дж., Осима, Дж., Моннат, Р. Дж., Мл. мутации WRN при синдроме Вернера. Гм. Мутат. 13: 271-279, 1999. Примечание: Опечатка: Hum. Мутат. 14: 84-85, 1999. [PubMed: 10220139] [Полный текст: https://doi.org/10.1002 / (SICI) 1098-1004 (1999) 13: 4 <271 :: AID-HUMU2> 3.0.CO; 2-Q]
Мотульский, А.Г., Шульц, А., Прист, Дж. Х. Синдром Вернера: хромосомы, гены и процесс старения. Lancet 279: 160–161, 1962. Примечание: Первоначально том I.
Накура, Дж., Вейсман, Э. М., Мики, Т., Камино, К., Ю., Ч.-Э., Осима, Дж., Фукути, К., Вебер, Дж. Л., Пиуссан, К., Мелараньо, М. И., Эпштейн, К. Дж., Скаппатиччи, С., Фраккаро, М., Мацумура, Т., Мурано, С., Йошида, С., Фудзивара, Ю., Сайда, Т., Огихара, Т., Мартин, Г. М., Шелленберг, Г. Д. Картирование гомозиготности локуса синдрома Вернера (WRN). Геномика 23: 600-608, 1994. [PubMed: 7851888] [Полный текст: https: // linkinghub.elsevier.com/retrieve/pii/S0888754384715485]
Норденсон, И. Хромосомные разрывы при синдроме Вернера и их предотвращение in vitro ферментами, улавливающими радикалы. Наследие 87: 151-154, 1977. [PubMed: 608842] [Полный текст: https://onlinelibrary.wiley.com/resolve/openurl?genre=article&sid=nlm:pubmed&issn=0018-0661&date=1977&volume=87&issue=2&spage=151]
Огбурн, К.Э., Осима, Дж., Пут, М., Чен, Р., Хант, К. Э., Голлахон, К. А., Рабинович, П. С., Мартин, Г. М. Генотоксин, индуцирующий апоптоз, отличает гетерозиготных носителей мутаций геликазы Вернера от мутантов дикого типа и гомозиготных мутантов. Гм. Genet. 101: 121-125, 1997. [PubMed: 9402954] [Полный текст: https: // dx.doi.org/10.1007/s0043
599]
Осима, Дж., Ю, К.-Э., Пиуссан, К., Кляйн, Г., Ябковски, Дж., Бальчи, С., Мики, Т., Накура, Дж., Огихара, Т., Эллс, Дж., Смит, МАК, Мелараньо, М.И., Фраккаро, М., Скаппатиччи, С., Мэтьюз, Дж., Уаис, С., Ярзебович, А., Шелленберг, Г.Д., Мартин, GM Гомозиготные и сложные гетерозиготные мутации в локусе синдрома Вернера. Гм. Molec. Genet. 5: 1909-1913, 1996. [PubMed: 8968742] [Полный текст: https: // академ.oup.com/hmg/article-lookup/doi/10.1093/hmg/5.12.1909]
Принс, П. Р., Огбурн, К. Э., Мозер, М. Дж., Эмонд, М. Дж., Мартин, Г.М., Моннат, Р. Дж., Мл. Слияние клеток корректирует чувствительность к 4-нитрохинолин-1-оксиду клеточных линий фибробластов синдрома Вернера. Гм. Genet. 105: 132-138, 1999. [PubMed: 10480367] [Полный текст: https: // dx.doi.org/10.1007/s0043998]
Раббиози, Г., Боррони, Г. Синдром Вернера: семь случаев в одной семье. Dermatologica 158: 355-360, 1979. [PubMed: 437224] [Полный текст: https://dx.doi.org/10.1159/000250780]
Рупрехт, К.W. Офтальмологические аспекты у пациентов с синдромом Вернера. Arch. Геронтол. Гериат. 9: 263-270, 1989. [PubMed: 2640084] [Полный текст: https: // linkinghub.elsevier.com/retrieve/pii/0167494389
Садакане Ю., Маэда К., Курода Ю., Хори К. Идентификация мутаций в мРНК ДНК-полимеразы бета от пациентов с синдромом Вернера. Biochem. Биофиз. Res. Commun. 200: 219-225, 1994. [PubMed: 7545922] [Полный текст: https://linkinghub.elsevier.com/retrieve/pii/S0006-291X(84)71437-9]
Солк, Д., Ау, К., Хоэн, Х., Мартин, Г. М. Влияние ферментов, улавливающих радикалы, и сниженное воздействие кислорода на рост и хромосомные аномалии культивированных фибробластов кожи при синдроме Вернера. Гм. Genet. 57: 269-275, 1981. [PubMed: 7250969] [Полный текст: https: // dx.doi.org/10.1007/BF00278942]
Солк, Д., Брайант, Э., Ау, К., Хон, Х., Мартин, Г. М. Систематические исследования роста, сокультивирования и исследования гибридизации клеток культивированных фибробластов кожи с синдромом Вернера. Гм. Genet. 58: 310-316, 1981. [PubMed: 7327553] [Полный текст: https://dx.doi.org/10.1007/BF00294930]
Солк, Д. Синдром Вернера: обзор недавних исследований с анализом метаболизма соединительной ткани, контроля роста культивируемых клеток и хромосомных аберраций. Гм. Genet. 62: 1-15, 1982. [PubMed: 6759366] [Полный текст: https: // dx.doi.org/10.1007/BF00295598]
Самантрей, С. К., Самантрей, С., Джонсон, С. К., Бхактавизиам, А. Синдром Вернера. Aust. New Zeal. J. Med. 7: 309-311, 1977. [PubMed: 269694] [Полный текст: https://onlinelibrary.wiley.com/resolve/openurl?genre=article&sid=nlm:pubmed&issn=0004-8291&date=1977&volume=7&issue=3&spage=309]
Скаппатиччи, С., Церимеле, Д., Фраккаро, М. Клональные структурные хромосомные перестройки в культурах первичных фибробластов и в лимфоцитах пациентов с синдромом Вернера. Гм. Genet. 62: 16-24, 1982. [PubMed: 7152523] [Полный текст: https: // dx.doi.org/10.1007/BF00295599]
Скаппатиччи, С., Форабоско, А., Боррони, Г., Ореккья, Г., Фраккаро, М. Клональные структурные хромосомные перестройки в лимфоцитах четырех пациентов с синдромом Вернера. Анна. Genet. 33: 5-8, 1990. [PubMed: 2369072]
Шелленберг, Г.Д., Мартин, Г. М., Вейсман, Э. М., Накура, Дж., Мики, Т., Огихара, Т. Картирование гомозиготности и синдром Вернера. (Письмо) Ланцет 339: 1002, 1992. [PubMed: 1348795] [Полный текст: https: // linkinghub.elsevier.com/retrieve/pii/0140-6736(92)
-5]
Шенберг, С., Нирмейер, М.Ф., Бутсма, Д., Хендерсон, Э., Герман, Дж. Синдром Вернера: пролиферация in vitro клонов клеток, несущих хромосомные транслокации. Am. J. Hum. Genet. 36: 387-397, 1984. [PubMed: 6324581]
Томас, В., Рубинштейн, М., Гото, М., Драйна, Д. Генетический анализ области синдрома Вернера на хромосоме 8p человека. Геномика 16: 685-690, 1993. [PubMed: 8325642] [Полный текст: https: // linkinghub.elsevier.com/retrieve/pii/S0888-7543(83)71248-6]
Твитт, Р., Гольдштейн, С. Синдром Вернера и биологическое старение: молекулярно-генетическая гипотеза. BioEssays 15: 421-426, 1993. [PubMed: 8357345] [Полный текст: https://doi.org/10.1002/bies.950150609]
Три, Т.Б., Комбс, Д. Т. Застойная кардиомиопатия при синдроме Вернера. (Письмо) Lancet 311: 1052-1053, 1978. Примечание: Первоначально том I. [PubMed: 76976] [Полный текст: https: // linkinghub.elsevier.com/retrieve/pii/S0140-6736(78)-9]
Уилли, Ф. С., Джонс, К. Дж., Скиннер, Дж. У., Хотон, М. Ф., Уоллис, К., Уинфорд-Томас, Д., Фарагер, Р. Г. А., Киплинг, Д. Теломераза предотвращает ускоренное старение клеток фибробластов синдрома Вернера. (Письмо) Nature Genet. 24: 16-17, 2000. [PubMed: 10615119] [Полный текст: https: // doi.org / 10.1038 / 71630]
Йе, Л., Накура, Дж., Мицуда, Н., Фудзиока, Ю., Камино, К., Охта, Т., Джинно, Ю., Ниикава, Н., Мики, Т., Огихара, Т. Генетическая ассоциация между микросателлитом хромосомы 8 (MS8-134) и синдромом Вернера (WRN): микродиссекция хромосомы и картирование гомозиготности. Геномика 28: 566-569, 1995. [PubMed: 74
] [Полный текст: https: // linkinghub.elsevier.com/retrieve/pii/S0888-7543(85)71189-5]Yu, C.-E., Oshima, J., Fu, Y.-H., Wijsman, E.M., Hisama, F., Alisch, R., Мэтьюз, С., Накура, Дж., Мики, Т., Куэйс, С., Мартин, Г. М., Маллиган, Дж., Шелленберг, Г. Д. Позиционное клонирование гена синдрома Вернера. Science 272: 258-262, 1996. [PubMed: 8602509] [Полный текст: https: // www.sciencemag.org/cgi/pmidlookup?view=long&pmid=8602509]
Ю, Ч.-Э., Осима, Дж., Годдард, К.А.Б., Мики, Т., Накура, Дж., Огихара, Т., Пут, М., Хоэн, Х., Фраккаро, М., Пиуссан, К., Мартин, Г. М., Шелленберг, Г. Д., Вейсман, Э. М. Неравновесие сцепления и исследования гаплотипов маркеров хромосомы 8p11.1-21.1 и синдрома Вернера. Am. J. Hum. Genet. 55: 356-364, 1994. [PubMed: 8037212]
Ю, С.-Э., Осима, Дж., Вейсман, Э.М., Накура, Дж., Мики, Т., Пиуссан, К., Мэтьюз, С., Фу, Ю.-Х., Маллиган, Дж., Мартин, Г.М., Schellenberg, GD, Совместная группа по синдрому Вернера. Мутации в консенсусных геликазных доменах гена синдрома Вернера. Am. J. Hum. Genet. 60: 330-341, 1997. [PubMed:
Синдром Вернера: обзор недавних исследований с анализом метаболизма соединительной ткани, контроля роста культивируемых клеток и хромосомных аберраций
Альберти KGMM, Young JDH, Hockaday TDR (1974) Синдром Вернера: метаболические наблюдения.Proc Roy Soc Med 67: 36–38
Google ученый
Angel M, Cano JF, del Olma JA, Villardell E (1980) Эндокринологические аспекты синдрома Вернера. Med Clin (Barc) 74: 317–321
Google ученый
Aram H, Fatourechi V (1974) Синдром Вернера: первые зарегистрированные случаи в Иране. Кутис 14: 215–218
Google ученый
Arlett CF, Harcourt SA (1980) Исследование радиочувствительности различных штаммов человеческих клеток.Cancer Res 40: 926–932
Google ученый
Aso K, Kondo S, Amano M (1980) Был продемонстрирован случай синдрома Вернера, в котором было продемонстрировано снижение активности тромбоцитарного сывороточного фактора для роста клеток и снижение потенциала роста фибробластов и эпидермальных клеток. Ниппон Хифука Гаккай Засси 90: 929–934
Google ученый
Аткин Н.Б., Бейкер М.С. (1978) Аномальные хромосомы и варианты гетерохроматина номер 1, обнаруженные в препаратах с С-диапазоном из 13 карцином мочевого пузыря.Cytobios 18: 101–109
Google ученый
Bartram CR, Koske-Westphal T, Passarge E (1976) Хроматидные обмены при атаксии, телеангиэктазии, синдроме Блума, синдроме Вернера и пигментной ксеродермии. Энн Хам Генет 40: 79–86
Google ученый
Basler JW, David JD, Agris PF (1979) Ухудшение синтеза коллагена и ультраструктура клеток, сопровождающая старение нормальных человеческих штаммов фибробластов и штаммов фибробластов синдрома Вернера.Exp Cell Res 118: 73–84
Google ученый
Beadle GF, Mackay IR, Whittingham S, Taggert G, Harris AW, Harrison LC (1978) Синдром Вернера, модель преждевременного старения? J Med 9: 377–403
Google ученый
Bertoni G, Lostia A (1967) Sulla sindrome di Werner (Contributo Clinico ed istologico). Ann Ottalmologia e Clinica Oculistica 93: 1130–1144
Google ученый
Bjornberg A (1976) Синдром Вернера и злокачественные новообразования.Acta Derm Venereol (Stockh) 56: 149–150
Google ученый
Blohme G, Smith U (1979) Метаболические исследования в случае синдрома Вернера. Диабете Метаб 5: 119–124
Google ученый
Brown WT (1979) Мутации человека, влияющие на старение — обзор. Механическое старение Dev 9: 325–336
Google ученый
Brown WT, Darlington GJ (1980) Термолабильные ферменты при прогерии и синдроме Вернера: доказательства, противоречащие гипотезе ошибки белка.Am J Hum Genet 32: 614–619
Google ученый
Brown WT, Darlington GJ, Arnold A, Fotino M (1980) Обнаружение антигенов HLA на фибробластах синдрома прогерии. Clin Genet 17: 213–219
Google ученый
Бакли CE, Roseman JM (1976) Иммунитет и выживание. J Am Geriatr Soc 24: 241–248
Google ученый
Бернет FM (1970) Иммунологический подход к старению.Ланцет 2: 358–360
Google ученый
Cerimele D, Cottoni F, Scappaticci S, Rabbiosi, Borroni G, Sanna E, Zei G, Fraccaro M (1982) Высокая распространенность синдрома Вернера на Сардинии. Описание шести пациентов и оценка частоты гена. Hum Genet
Corrales Hernandez JJ, Miralles Garcia JM, Rodriguez Commes JL, Martin Vasallo P, Garcia Diez LC, de Pablo Davila F (1980) Новые ферментативные и эндрокринологические аспекты в двух случаях синдрома Вернера.Med Clin (Barc) 74: 355–360
Google ученый
Darlington GJ, Dutkowski R, Brown WT (1981) Частоты обмена сестринских хроматид у пациентов с прогерией и синдромом Вернера. Am J Hum Genet 33: 762–766
Google ученый
Djawari D, Lukaschek E, Jecht E (1980) Измененный клеточный иммунитет при синдроме Вернера. Dermatologica 161: 233–237
Google ученый
Elliott RJ, Gardner DL (1979) Изменения гликозаминогликанов суставного хряща человека, связанные с ege.Ann Rheum Dis: 233–237
Эпштейн К.Дж., Мартин Г.М., Шульц А.Л., Мотульский А.Г. (1966) Синдром Вернера: обзор его симптоматологии, естественного течения, патологических особенностей, генетики и связи с естественным процессом старения. Медицина 45: 177–221
Google ученый
Fellini SA, Pacifici M, Holtzer H (1981) Изменения сульфатированных протеогликанов, синтезируемых «стареющими» хондроцитами. II. Позвоночные столбы, культивируемые органами.J Biol Chem 256: 1038–1043
Google ученый
Field JB, Loube SD (1960) Наблюдения за сахарным диабетом, связанным с синдромом Вернера. Метаболизм 9: 118–124
Google ученый
Фитцджеральд PH (1975) Механизм анеуплоидии Х-хромосомы в лимфоцитах стареющих женщин. Хум Генет 28: 153–158
Google ученый
Fleischmajer R, Nedwich A (1973) Синдром Вернера.Am J Med 54: 111–118
Google ученый
Fujiwara Y, Higashikawa T, Tatsumi M (1977) Замедленная скорость репликации ДНК и нормальный уровень репарации ДНК в фибробластах синдрома Вернера в культуре. J Cell Physiol 92: 365–374
Google ученый
Галлоуэй С.М., Бактон К.Э. (1978) Анеуплоидия и старение: хромосомные исследования на случайной выборке населения с использованием G-диапазона.Cytogenet Cell Genet 20: 78–95
Google ученый
German J (1972) Гены, которые увеличивают хромосомную нестабильность в соматических клетках и предрасполагают к раку. Prog Med Genet 8: 61–101
Google ученый
Gershon D (1979) Текущее состояние возрастных ферментов: альтернативные механизмы. Механическое старение Dev 9: 189–196
Google ученый
Гиббс Д. Д. (1967) Синдром Вернера («прогерия взрослого»).Proc R Soc Med 60: 135–136
Google ученый
Goldstein S (1979) Исследования возрастных заболеваний в культивируемых фибробластах кожи. J Invest Dermatol 73: 19–23
Google ученый
Goldstein S, Harley CB (1979) Исследования заболеваний, связанных с возрастом, in vitro. Fed Proc 38: 1862–1867
Google ученый
Goldstein S, Moerman EJ (1975) Термолабильные ферменты в фибробластах синдрома Вернера.Природа 255: 159
Google ученый
Goldstein S, Moerman EJ (1976) Дефектные белки в нормальных и аномальных фибробластах человека во время старения in vitro. Междисциплинарные темы Геронт 10: 24–43
Google ученый
Goldstein S, Niewiarowski S (1976) Повышенная прокоагулянтная активность культивированных фибробластов от прогерии и синдрома преждевременного старения Вернера.Природа 260: 711–713
Google ученый
Goldstein S, Singal DP (1974) Изменение продуктов гена фибробластов in vitro у субъекта с синдромом Вернера. Nature 251: 719–721
Google ученый
Goldstein S, Stotland D, Cordeiro RAJ (1976) Снижение протеолиза и увеличение оттока аминокислот в стареющих фибробластах человека. Mech Aging Dev 5: 221–233
Google ученый
Goto M, Murata K (1978) Экскреция с мочой макромолекулярных кислых гликозаминогликанов при синдроме Вернера.Clin Chim Acta 85: 101–106
Google ученый
Goto M, Horiuchi Y, Tanimoto K, Ishii T., Nakashima H (1978) Синдром Вернера: анализ 15 случаев с обзором японской литературы. J Am Geriatr Soc 26: 341–347
Google ученый
Goto M, Horiuchi Y, Okumura K, Tada T, Kawata M, Ohmori K (1979) Иммунологические аномалии старения: анализ субпопуляций T-лимфоцитов синдрома Вернера.J Clin Invest 64: 695–699
Google ученый
Goto M, Tanimoto K, Horiuchi Y, Sasazuji T (1981) Семейный анализ синдрома Вернера: обзор 42 японских семей с обзором литературы. Clin Genet 19: 8–15
Google ученый
Gottesman T, Zala L, Vogel A, Mumenthaler M (1980) Das Werner-Syndrom. Schweiz Med Wochenschr 110: 246–250
Google ученый
Gupta S (1981) Субпопуляции Т-лимфоцитов человека.Геронтология 27: 181–186
Google ученый
Холл Д.А. (1981) Геронтология: коллагеновая болезнь. Clin Endocrinol Metab 10: 23–55
Google ученый
Harley CB, Pollard JW, Chamberlain JW, Stanners CP, Goldstein S (1980) Ошибки синтеза белка не увеличиваются при старении культивируемых фибробластов человека. Proc Natl Acad Sci USA 77: 1885–1889
Google ученый
Harnden DG, Benn PA, Oxford JM, Taylor AMR, Webb TP (1976) Цитогенетически маркированные клоны в человеческих фибробластах, культивируемых от нормальных субъектов.Somatic Cell Genet 2: 55–62
Google ученый
Хигасикава Т., Фудзивара Ю. (1978) Нормальный уровень внепланового синтеза ДНК в фибробластах синдрома Вернера в культуре. Exp Cell Res 133: 438–442
Google ученый
Hoehn H, Bryant EM, Au K, Norwood TH, Boman H, Martin GM (1975) Пестрый транслокационный мозаицизм в культурах фибробластов кожи человека. Cytogenet Cell Genet 15: 282–298
Google ученый
Холлидей Р., Таррант Г.М. (1972) Измененные ферменты в стареющих фибробластах человека.Природа 238: 26–30
Google ученый
Холлидей Р., Портерфилд Дж. С., Гиббс Д. Д. (1974) Преждевременное старение и появление измененного фермента в фибробластах синдрома Вернера. Природа 248: 762–763
Google ученый
Hoppe W, Koritsch H-D (1972) Ein weiterer Fall von Meningeom bei einem Patienten mit Werner-Syndrom. Psychiatr Neurol Med Psychol (Лейпц) 24: 611–617
Google ученый
Houben A, Houbion A, Remacle J (1980) Лизосомные и митохондриальные термолабильные ферменты в фибробиастах синдрома Вернера.Опыт Геронтол 15: 629–631
Google ученый
Храбко Р.П., Милгром Х., Шварц Р.А. (1982) Синдром Вернера с ассоциированными злокачественными новообразованиями. Arch Dermatol 118: 106–108
Google ученый
Ishii T, Hosoda Y (1975) Синдром Вернера: отчет о вскрытии одного случая с обзором патологических данных, описанных в литературе. J Am Geriatr Soc 23: 145–154
Google ученый
Кан А., Гийозо А., Коттро Д., Мари Дж., Бурель М., Бойвин П., Дрейфус Дж. К. (1977) Точность синтеза белка и старение in vitro: поиск измененных ферментов в стареющих культивируемых клетках печени человека.Геронтология 23: 174–184
Google ученый
Кобаяши С., Гибо Х, Сугита К., Комия И., Ямада Т. (1980) Синдром Вернера, связанный с менингиомой. Нейрохирургия 7: 517–520
Google ученый
Littlefield LG, Mailhes JB (1975) Наблюдения за клонами de novo цитогенетически аберрантных клеток в штаммах первичных фибробластов от фенотипически нормальных женщин.Am J Hum Genet 27: 190–197
Google ученый
Макинодан Т., Хейдрик М.Л., Нордин А.А. (1975) Иммунодефицит и аутоиммунитет при старении. Врожденные пороки 11: 193–198
Google ученый
Marklund S, Nordensson I, Back O (1981) Нормальная супероксиддисмутаза CuZn, каталаза и глутатионпероксидаза при синдроме Вернера. J Gerontol 36: 405–409
Google ученый
Мартин Г.М. (1978) Генетические синдромы у человека, потенциально имеющие отношение к патобиологии старения.Врожденные пороки 14: 5–39
Google ученый
Martin GM, Sprague CA, Epstein CJ (1970) Продолжительность репликативной жизни культивируемых человеческих клеток: влияние возраста донора, ткани и генотипа. Lab Invest 23: 86–91
Google ученый
Matuoka K, Mitsui Y (1981a) Изменения гликозаминогликанов клеточной поверхности в диплоидных фибробластах человека во время старения in vitro. Mech Aging Dev 15: 153–163
Google ученый
Matuoka K, Mitsui Y (1981b) Вовлечение гепарансульфата клеточной поверхности в зависимое от плотности ингибирование пролиферации клеток.Функция структуры ячеек 6: 23–33
Google ученый
Mourao PAS, Machado-Santelli GM (1978) Сульфатированные гликозаминогликаны клеток, выращенных в культуре: исчезновение дерматансульфата в последующих субкультурах фибробластов. Cell Differ 7: 367–374
Google ученый
Мурата К. (1982) Кислые гликозаминогликаны мочи при синдроме Вернера. Experientia 38: 313–314
Google ученый
Мурата К., Накашима Х (1982) Синдром Вернера: 24 случая с обзором японской медицинской литературы.J Am Geriatr Soc 30: 303–308
Google ученый
Nakao Y, Kishihara M, Yoshimi H, Inoue Y, Tanaka K, Sakamoto N, Matsukura S, Imura H, Ichihashi M, Fujiwara Y (1978) Синдром Вернера: характеристики in vivo и in vitro как модель старения . Am J Med 65: 919–932
Google ученый
Nakao Y, Hattori T, Takasuki K, Kuroda Y, Nakaji T., Fujiwara Y, Kishihara M, Baba Y, Fujita T (1980) Иммунологические исследования синдрома Вернера.Clin Exp Immunol 42: 10–19
Google ученый
Nordenson I (1977) Хромосомные разрывы при синдроме Вернера и их предотвращение in vitro ферментами, улавливающими радикалы. Наследие 87: 151–154
Google ученый
Norwood TH, Pendergrass WR, Sprague CA, Martin GM (1974) Преобладание сенесцентного фенотипа в гетерокарионах между репликативными и пострепликативными человеческими фибробластоподобными клетками.Proc Natl Acad Sci USA 71: 2231–2235
Google ученый
Norwood TH, Pendergrass WR, Martin GM (1975) Повторная инициация синтеза ДНК в стареющих фибробластах человека после слияния с клетками с неограниченным потенциалом роста. J Cell Biol 64: 551–556
Google ученый
Norwood TH, Hoehn H, Salk D, Martin GM (1979) Клеточное старение при синдроме Вернера: уникальный фенотип? J Invest Dermatol 73: 92–96
Google ученый
Охира А., Ногами Х (1980) Возрастные изменения физических и химических свойств протеогликанов, синтезируемых реберными и матриксными хрящами у крыс.J Biol Chem 255: 1346–1350
Google ученый
Оппенгеймер Б.С., Кугель В.Х. (1934) Синдром Вернера — наследственное семейное заболевание со склеродермией, двусторонней ювенильной катарактой, преждевременным поседением волос и эндокринной стигматизацией. Trans Assoc Am Physitors 49: 358
Google ученый
Оппенгеймер Б.С., Кугель В.Х. (1941) Синдром Вернера: отчет о первом вскрытии трупа и результаты нового случая.Am J Med Sci 202: 629
Google ученый
Orgel LE (1963) Поддержание точности синтеза белка и его влияние на старение. Proc Natl Acad Sci USA 49: 517–521
Google ученый
Orgel LE (1973) Старение клонов клеток млекопитающих. Nature 243: 441–445
Google ученый
Pendergrass WR, Martin GM, Bornstein P (1976) Доказательства, противоречащие гипотезе белковой ошибки для старения in vitro.J Cell Physiol 87: 3–14
Google ученый
Rabbiosi G, Borroni G (1979) Синдром Вернера: семь случаев в одной семье. Dermatologica 158: 355–360
Google ученый
Reed R, Seville RH, Tattersall RN (1953) Синдром Вернера. Br J Dermatol 65: 165–176
Google ученый
Райли Т.Р., Виланд Р.Г., Маркис Дж., Хамви С.Дж. (1965) Синдром Вернера.Ann Intern Med 63: 285–294
Google ученый
Роберт Л. (1981) Старение соединительной ткани. Experientia 37: 1055–1058
Google ученый
Rosen RS, Cimini R, Coblentz D (1970) Синдром Вернера. Br J Radiol 43: 193–198
Google ученый
Rothstein M (1979) Образование измененных ферментов у стареющих животных.Механическое старение Dev 9: 197–202
Google ученый
Rowley JD (1977) Неслучайные хромосомные изменения в злокачественных клетках человека. В: Sparkes RS, Comings DE, Fox CF (eds) Molecular Human cytogenetics. Academic Press, New York, pp 457–472
Google ученый
Rowley JD (1982) Идентификация константных участков хромосом, участвующих в гематологических злокачественных заболеваниях человека.Наука 216: 749–751
Google ученый
Саламон Р., Богданович Б., Лазович-Тепавак О., Блажевич А., Маканович-Богнер К. (1978) Синдром Вернера и клеточные иммунные реакции. Acta Derm Venereol (Stockh) 58: 543–544
Google ученый
Salk D, Bryant E, Au K, Hoehn H, Martin GM (1981a) Систематические исследования роста, сокультивирования и исследования гибридизации клеток культивированных фибробластов кожи при синдроме Вернера.Хум Генет 58: 310–316
Google ученый
Salk D, Au K, Hoehn H, Martin GM (1981b) Цитогенетика культивированных фибробластов кожи с синдромом Вернера: мозаицизм с разнообразной транслокацией. Cytogenet Cell Genet 30: 92–107
Google ученый
Salk D, Au K, Hoehn H, Martin GM (1981c) Влияние ферментов, улавливающих радикалы, и пониженное воздействие кислорода на рост и хромосомные аномалии культивированных фибробластов кожи при синдроме Вернера.Хум Генет 57: 269–275
Google ученый
Salk D, Au K, Hoehn H, Stenchever MR, Martin GM (1981d) Доказательства клонального ослабления, клональной преемственности и клональной экспансии в массовых культурах стареющих фибробластов кожи с синдромом Вернера. Cytogenet Cell Genet 30: 108–117
Google ученый
Salk D, Maxwell C, Au K, Hoehn H, Martin GM (1981e) Эволюция и клональность стабильных хромосомных перестроек при синдромах Блума и Вернера.Am J Hum Genet 33: 119A
Google ученый
Salk D, Carlson C, Martin GM (1981f) Ядерная морфология старения и сывороточного голодания нормальных и культивированных фибробластов кожи с синдромом Вернера. Геронтолог 21:89
Google ученый
Salk D, Au K, Hoehn H, Martin GM (будет опубликовано) Соматические хромосомные перестройки происходят in vivo и in vitro при синдроме Вернера (прогерия у взрослых)
Scappaticci S, Cerimele D, Fraccaro M (1982) ) Клональные структурные хромосомные перестройки в первичных культурах фибробластов и в лимфоцитах пациентов с синдромом Вернера.Хум Генет 62: 16–24
Google ученый
Schachtschabel DO, Wever J (1978) Связанное с возрастом снижение синтеза гликозаминогликанов культивированными фибробластами человека (WI-38). Механическое старение Dev 8: 257–264
Google ученый
Schonberg SA, Henderson E, Niermeijer MF, German J (1981) Синдром Вернера: преимущественное размножение клонов с транслокациями. Am J Hum Genet 33: 120A
Google ученый
Schonberg S, Henderson E, Niermeijer MF, Bootsma D, German J (будет опубликовано) Синдром Вернера: преимущественная пролиферация клонов с транслокациями хромосом
Shakespeare V, Buchanan JH (1978) Исследования по изоме фосфоглюкозы культивированные человеческие фибробласты: отсутствие заметных эффектов старения на ферменты.J Cell Physiol 94: 105–116
Google ученый
Sluke G, Schachtschabel DO, Wever J (1981) Возрастные изменения в структуре распределения гликозаминогликанов, синтезируемых культивированными диплоидными фибробластами человека (WI-38). Механическое старение Dev 16: 19–27
Google ученый
Smith JC, Singh JP, Lillquist JS, Goon DS, Stiles CD (1982) Факторы роста, прикрепленные к клеточному субстрату, являются митогенно активными in situ.Природа 296: 154–156
Google ученый
Smith RC, Winer LH, Martel S (1955) Синдром Вернера: отчет о двух случаях. Arch Dermatol 71: 197–204
Google ученый
Smith U, Digirolamo M, Blohme G, Kral JG, Tisell L-E (1980) Возможные системные метаболические эффекты регионарного ожирения у пациента с синдромом Вернера. Int J Obes 4: 153–163
Google ученый
Subino S, Fiori PL, Lubinu G, Scappaticci S, Cerimele D (1982) Иммунофлуоресценция тубулиновой системы в фибробластах кожи человека при синдроме Вернера, саркоме Капоши и псориазе.Arch Dermatol Res 272: 143–145
Google ученый
Sweet MBE, Thonar EJMA, Marsh J (1979) Возрастные изменения в структуре протеогликана. Arch Biochem Biophys 198: 439–448
Google ученый
Таджима Т., Иидзима К., Ватанабе Т. (1978) Синтез коллагена культивируемых фибробластов от синдрома преждевременного старения Вернера. Experientia 34: 1459–1460
Google ученый
Таджима Т., Ватанабе Т., Иидзима К., Охика Y, Ямагути Х. (1981) Увеличение синтеза и накопления гликозаминогликанов на поверхности клеток культивируемых фибробластов кожи при синдроме Вернера.Опыт Патол (Йена) 20: 221–229
Google ученый
Tanaka K, Nakazawa T, Okada Y, Kumahara Y (1979) Увеличение синтеза ДНК в клетках синдрома Вернера путем гибридизации с нормальными диплоидными клетками человека и клетками HeLa. Exp Cell Res 123: 261–267
Google ученый
Танака К., Накадзава Т., Окада Ю., Кумахара Ю. (1980) Роль ядерной и цитоплазматической среды в замедленном синтезе ДНК в клетках с синдромом Вернера.Exp Cell Res 127: 185–190
Google ученый
Таненбаум М.Х. (1965) Синдром Вернера: прогерия взрослого. Arch Intern Med 116: 499–504
Google ученый
Tao LC, Stecker E, Gardner HA (1971) Синдром Вернера и острый миелоидный лейкоз. Can Med Assoc J 105: 952–954
Google ученый
Thannhauser SJ (1945) Синдром Вернера (прогерия взрослого) и синдром Ротмунда: два типа тесно связанных наследственно-семейных атрофических дерматозов с ювенильными катарактами и эндокринными особенностями.Критическое исследование с пятью новыми случаями. Энн Интерн Мед 23: 559
Google ученый
Tokunaga M, Futami T, Wakamatsu E, Endo M, Yosizawa Z (1975) Синдром Вернера как «гиалуронурия». Clin Chim Acta 62: 89–96
Google ученый
Токунага М., Мори С., Сато К., Накамура К., Вакамацу Е. (1976) Патологоанатомическое исследование случая синдрома Вернера. J Am Geriatr Soc 24: 407–411
Google ученый
Tokunaga M, Wakamatsu E, Yosizawa Z (1979) Выделение глюкозосодержащих олигосахаридов в моче ортопедических пациентов.Tohoku J Exp Med 128: 71–79
Google ученый
Фогель К.Г., Кендалл В.П., Сапиен Р. (1981) Синтез и состав гликозаминогликанов в человеческих фибробластах во время клеточного старения in vitro (IMR-90). J Cell Physiol 107: 271–281
Google ученый
Wight TN (1980) Протеогликаны сосудов и тромбообразование. Prog Hemost Thromb 5: 1–39
Google ученый
Вернер О. (1904) Über Katarakt in Verbindung mit Skelerodermie (докторская диссертация, Кильский университет).Schmidt and Kaluning, Kiel
Wever J, Schachtschabel DO, Sluke G, Wever G (1980) Влияние краткосрочного или длительного лечения экзогенными гликозаминогликанами на рост и синтез гликозаминогликанов человеческих фибробластов (WI-38) в культуре . Механическое старение Dev 14: 89–99
Google ученый
Войтык Р.И., Гольдштейн С. (1980) Верность белкового синтеза не снижается при старении культивируемых фибробластов человека.J Cell Physiol 103: 299–303
Google ученый
Yatscoff RW, Goldstein S, Freeman KB (1978) Сохранение генов, кодирующих белки, синтезируемые в митохондриях человека. Somatic Cell Genet 4: 633–645
Google ученый
Юнис Э.Дж., Фернандес Г., Гринберг Л.Дж. (1976) Иммунология опухолей, аутоиммунитет и старение. J Am Geriatr Soc 24: 258–263
Google ученый
Zackai AH, Weber D, Noth R (1974) Кардиологические находки при синдроме Вернера.Гериатрия 29: 141–148
Google ученый
Залла Я.А. (1980) Синдром Вернера. Кутис 25: 275–278
Google ученый
-

 Промывание носа физраствором грудничку: польза, техника и меры предосторожности
Промывание носа физраствором грудничку: польза, техника и меры предосторожности - Отпадение пупка у новорожденных: сроки, уход и возможные осложнения
- Как сшить пеленки для новорожденного своими руками: пошаговая инструкция
- Развитие мелкой моторики у дошкольников: эффективные упражнения и игры
- Размеры пеленок для новорожденных: как выбрать оптимальный вариант
- Лечение насморка у новорожденных: эффективные методы и рекомендации педиатров