Передается ли олигофрения по наследству? — Авис
Передается ли олигофрения по наследству?
Вопрос: хочу взять девочку из детского дома 3-х лет. У ее мамы олигофрения в легкой форме. Передается ли это по наследству?
Варвара
Ответ:
Олигофрения относится к вопросам дефектологии, к психологии лишь отчасти, но мы постараемся дать информативный ответ. Давайте для начала разберемся, что вы знаете о самом диагнозе?
Олигофрения— врожденное или рано приобретенное (в первые 3 года жизни) слабоумие, которое выражается в недоразвитии всей психики, но преимущественно интеллекта. Если у матери олигофрения носит приобретенный характер (сильные травмы головы, осложнение тяжелого инф. заболевания и пр.) то такая форма не передается по наследству.
Если олигофрения врожденная, то вероятность наследования — достаточно высока. Также, немаловажную роль играет психический статус второго родителя (олигофрен или здоровый). Поэтому, если у вас есть возможность узнать полный анамнез матери ребенка, то это Вам поможет. К тому же, как правило, наследственные формы олигофрении имеют внешние признаки — диспластичность телосложения и пороки развития отдельных органов. Так что, если мама малыша внешне абсолютно нормально выглядит, то, скорее всего вероятность наследственной формы олигофрении у ребенка минимальна. Если ребенку уже три года, эти признаки внешне так же могут быть видны. Другой вопрос — как она, мать, вынашивала ребенка, которого вы хотите взять? Были ли там быть факторы, которые могли оказать негативное влияние на мозг ребенка (алкоголь, наркотики, какие-то инфекции)? В любом случае, если Вы такой информацией не обладаете, посоветуйтесь с грамотным психиатром, детским неврологом, возможно, Вам посоветуют провести генетическое обследование ребенка.
Вам выпало принять сложное решение, мы советуем выслушать мнения всех специалистов, но принимать решение так, как подскажет Вам сердце.
Не передать по наследству
В идеале сегодня каждая супружеская пара, планирующая детей (или партнеры, желающие иметь совместного ребенка), должна проходить медико-генетическую консультацию у специалистов – врачей-генетиков в женских консультациях, платных клиниках или НИИ генетики РАМН. Есть в стране и специализированные медико-генетические консультации. Ухудшающаяся экология, постоянные стрессы и прочие «прелести» жизни в современных мегаполисах не способствуют рождению здорового потомства и лишь увеличивают число врожденных патологий у детей. У любой супружеской пары риск рождения ребенка с пороками развития составляет 6%, поскольку каждый человек является носителем генных изменений и мутаций, передающихся из поколения в поколение, и багаж этот нарастает как снежный ком. Каждый из нас несет в своем хромосомном наборе в среднем 10–12 дефектных генов, и даже если вы и все ваши отцы, деды и прадеды до седьмого колена были здоровы, нет никакой гарантии, что именно с вашим наследником природа не сыграет злую шутку и дефект не проявит себя в виде какой-то патологии. Наиболее перспективными считаются новейшие методы диагностики – ДНК-диагностика и молекулярно-генетический анализ.
Кто в группе риска?
Но в большинстве случаев будущие родители пренебрегают визитом к генетику, надеясь на авось. Врачи оставляют принятие решения об обследовании на усмотрение мужчины и женщины, но далеко не всегда. Есть немало случаев, когда генетическая консультация назначается по медицинским показаниям: если у будущих родителей осложненный анамнез, то есть у отца или матери есть родственники, дети или сами они страдают каким-то наследственным заболеванием; если у женщины были 1–2 выкидыша; если будущие родители – дальние родственники; если женщина старше 35 лет; если мужчина старше 50 лет.
Если все-таки будущие родители по каким-то соображениям решили не консультироваться с генетиками, от дородовых обследований беременной женщине все же отказываться не стоит.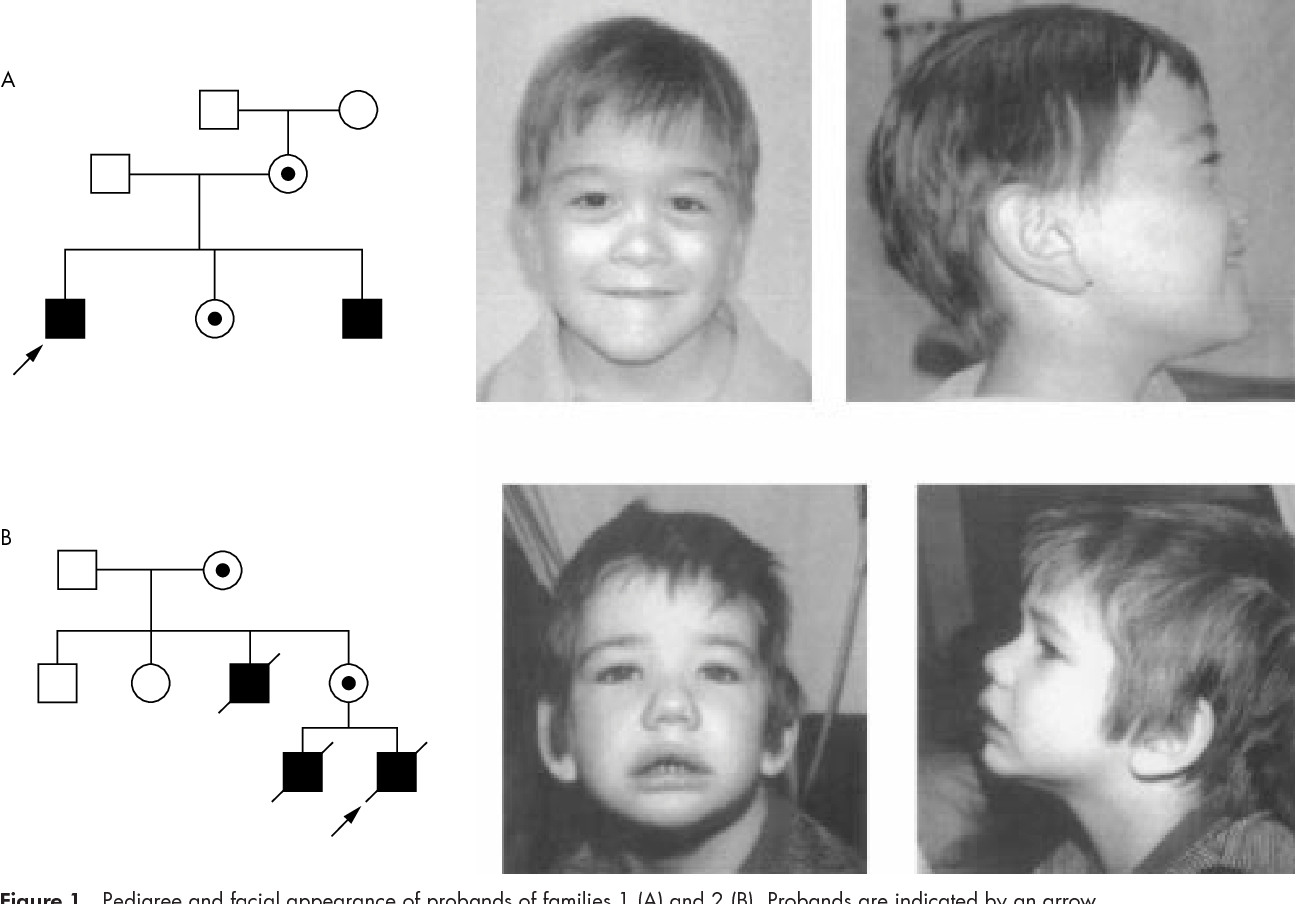 Ведь исследования делаются на 14–16-й неделях беременности, и если у плода выявятся дефекты развития, у родителей будет время для принятия решения на основе полученной информации – прерывать беременность или нет. Обследовав будущую маму, врачи смогут сказать, насколько серьезная патология у ребенка, как она отразится на его здоровье и вообще, жизнеспособным он родится или нет. Медики лишь сообщают информацию, но последнее слово всегда остается за женщиной и мужчиной.
Ведь исследования делаются на 14–16-й неделях беременности, и если у плода выявятся дефекты развития, у родителей будет время для принятия решения на основе полученной информации – прерывать беременность или нет. Обследовав будущую маму, врачи смогут сказать, насколько серьезная патология у ребенка, как она отразится на его здоровье и вообще, жизнеспособным он родится или нет. Медики лишь сообщают информацию, но последнее слово всегда остается за женщиной и мужчиной.
Методы дородовой диагностики наследственных болезней, давно и широко используемые на Западе, все активнее применяются и в России. Так, в соответствии с приказами Минздравсоцразвития РФ, сегодня каждая беременная женщина должна трижды пройти обследование на генетические дефекты плода, которое состоит из УЗИ, компьютерной томографии и исследования белково-сывороточных факторов. Эти виды диагностики позволяют выявить около 70% врожденных патологий, в том числе такие хромосомные заболевания, как болезнь Дауна или болезнь Эдвардса, предопределяющяя умственную неполноценность ребенка, пороки развития сердца, нервной трубки, головного мозга, аномальное развитие рук и ног. Что касается синдрома Дауна, то в настоящее время в России ведется постоянный скрининг по выявлению патологии – обследуют всех беременных россиянок, причем женщин старше 35 лет – дважды в обязательном порядке (известно, что чем старше беременная, тем выше вероятность болезни Дауна у ребенка). Дело в том, что синдром Дауна – одна из самых распространенных наследственных патологий: даунята рождаются с частотой 1 на 800 здоровых детей. Немало и других серьезных хронических заболеваний, которые передаются по наследству от матери. Например, инсулинозависимый сахарный диабет (первого типа) или бронхиальная астма.
Анализы первого дня
Младенцев также обследуют на наследственные и врожденные заболевания – не явные, а уже скрытые, которые невозможно выявить с помощью ультразвука и других дородовых диагностических методов. Например, в США всех новорожденных обследуют на 8 наследственных болезней, в России – на 5–6. Так, все малыши без исключения еще в роддоме сегодня проходят анализы на фенилкетонурию, врожденный гипотериоз, диагностику на адреногенитальный синдром, галактоземию и муковисцидоз. У каждого маленького человечка на пятый день жизни берут кровь и отправляют ее на генетический анализ, чтобы определить, нет ли повреждений в генах.
Так, все малыши без исключения еще в роддоме сегодня проходят анализы на фенилкетонурию, врожденный гипотериоз, диагностику на адреногенитальный синдром, галактоземию и муковисцидоз. У каждого маленького человечка на пятый день жизни берут кровь и отправляют ее на генетический анализ, чтобы определить, нет ли повреждений в генах.
Фенилкетонурия – не что иное, как олигофрения, умственная отсталость, развития которой при своевременной диагностике сегодня можно избежать. Фенилаланин – аминокислота, которая есть во всех белковых продуктах и которая в норме расщепляется и выводится из организма. Но если процесс метаболизма нарушен, аминокислота и токсины накапливаются в организме, что приводит к тяжелому поражению ЦНС, изменениям в мозгу и слабоумию. Чтобы этого не случилось, детей с непереносимостью фенилаланина уже с трех недель жизни переводят на специальную диету, и они нормально развиваются, растут, ходят в обычные детские сады, школы и вырастают здоровыми, полноценными людьми, а не идиотами. Казалось бы, всего лишь диета – дети до 12–18 лет получают продукты, содержащие белок, освобожденный от пагубной аминокислоты, – но если ее не соблюдать, ребенок обречен на умственную отсталость. Каждый год в России выявляют около 150 детей, не переносящих фенилаланин, и порядка 450 детей, организм которых (вернее, щитовидная железа) не вырабатывает гормон тироксин. При своевременно выявленной врожденной патологии всем им врачи могут помочь.
Вирус-убийца: найти и обезвредить
Есть еще одна проблема, с которой сталкиваются будущие мамы и на которую не стоит закрывать глаза, – хронические инфекции половых органов. Еще на этапе планирования беременности и женщина, и ее партнер должны сдать комплексный анализ на инфекции. В него входят цитомегаловирус, токсоплазмоз, хламидиоз, герпетическая и урогенитальные инфекции. Какие-то из них, если нет обострения, не опасны для ребенка, а какие-то надо обязательно сначала лечить, а потом уже беременеть. Например, хламидиоз или герпес. Особенно опасна последняя: если женщина заражается герпесом во время беременности, очень велика степень рождения ребенка с очень серьезными проблемами со здоровьем или что беременность закончится выкидышем. Герпетическая инфекция существует двух типов: первый поражает губы, носогубный треугольник, глаза, шею и руки, второй – гениталии и ягодицы. Именно второй тип очень опасен для беременной и ее ребенка: вирус может стать причиной тяжелейших врожденных заболеваний – эпилепсии, слепоты, глухоты, детского церебрального паралича. «Герпес убивает мозг», – говорят врачи, и если будущая мать заражается генитальным герпесом, нередко ставится вопрос о прерывании беременности – слишком уж велик риск рождения неполноценного младенца.
Особенно опасна последняя: если женщина заражается герпесом во время беременности, очень велика степень рождения ребенка с очень серьезными проблемами со здоровьем или что беременность закончится выкидышем. Герпетическая инфекция существует двух типов: первый поражает губы, носогубный треугольник, глаза, шею и руки, второй – гениталии и ягодицы. Именно второй тип очень опасен для беременной и ее ребенка: вирус может стать причиной тяжелейших врожденных заболеваний – эпилепсии, слепоты, глухоты, детского церебрального паралича. «Герпес убивает мозг», – говорят врачи, и если будущая мать заражается генитальным герпесом, нередко ставится вопрос о прерывании беременности – слишком уж велик риск рождения неполноценного младенца.
симптомы, лечение, причины болезни, первые признаки
Описание
Олигофрения – это недоразвитие психологических функций из-за воздействия неблагоприятных факторов во время внутриутробного развития либо после рождения.
Выделяют три степени олигофрении: идиотия, имбецильность, дебильность.
Идиотия — самая тяжёлая степень недоразвития интеллекта. Пациенты с идиотией не способны к самообслуживанию, выражают только примитивные эмоции (крик, плач), не способны узнавать родных и близких, некоторые не способны ходить, совершают лишь стереотипные движения, такие как покачивания вперёд-назад, существует тенденция к копированию эмоций и мимики окружающих пациента людей. Такие пациенты из-за нужды в постоянном контроле и уходе содержатся в психоневрологических интернатах и иных специальных учреждениях соцобеспечения. Как правило, пациенты с идиотией не доживают до периода зрелости, т.к. помимо психологических отклонений этому заболеванию сопутствуют множественные дефекты внутренних органов, в результате чего иммунная система несовершенна. В итоге организм не способен регулировать свои соматические функции, что приводит к снижению его защитных свойств. Такие пациенты чаще погибают от интеркуррентных заболеваний (острые заболевания, которые развиваются на фоне имеющегося хронического заболевания).![]()
Имбецильность – это одна из форм олигофрении, характеризующаяся тяжёлой умственной отсталостью и неспособностью пациента к абстрактному мышлению. У человека с имбецильностью плохая артикуляция, наблюдаются заикания. Словарный запас скуден, что мешает пациенту строить длинные сложные фразы. Больной с диагнозом имбецильность способен выучить буквы, но проводить разбор слова по буквам или выстраивать из букв слово не может. Пациент правильно называет предметы, знает, как эти предметы использовать. Больной часто бывает привязан к своим близким. Пациенты способны обучиться ухаживать за собой: надевать и снимать одежду, есть ложкой, ходить в туалет, проводить гигиенические мероприятия. Человек с имбецильностью способен повторять простые трудовые действия за окружающими людьми, но без пристального контроля пациент быстро начинает отвлекаться и перестаёт работать. При должных занятиях возможна его адаптация к узкому кругу людей, также возможно обучение лёгкому труду. Как правило, люди с диагнозом имбецильность живут в своих семьях.
Дебильность – это умственная отсталость лёгкой степени с тенденцией к конкретно-ситуационному мышлению. Пациенты с диагнозом дебильность характеризуются плохой памятью, нормальным словарным запасом, высокой способностью к подражанию, полной сохранностью моторных функций, хорошей внятной речью. Больной человек прекрасно повторяет за окружающими их действия, способен к физическому труду, обучается счёту. Однако такие люди лишены понимания их действий, то есть пациент может помыть посуду без моющего средства, добавить в суп неочищенную курицу, купить абсолютно ненужную вещь в магазине, пойти в школу за несколько часов до начала занятий. Обучение в обычном классе для пациента с диагнозом дебильность неэффективно по причине отсутствия абстрактного мышления. В специализированных классах ребёнок может обучиться письму, чтению, простым математическим вычислениям (вычитание, сложение, умножение, деление), получить базовые знания об окружающем мире.
По вызвавшему фактору выделяют следующие типы олигофрении:
- Эндогенные, генетические: к ним относятся хромосомные абберации (синдром Дауна), наследственные недостаточности ферментов (фенилкетонурия, синдром Марфана, галактоземия), сочетанные поражения центральной нервной и костной систем.
- Эмбриопатии и фетопатии (fetus – плод): перенесённые инфекционные заболевания во время беременности (краснуха, сифилис, токсоплазмоз), приём алкоголя и нерекомендуемых лекарственных средств в период вынашивания.
- Родовые травмы, асфиксия при родах, перенесённые тяжелые инфекции в раннем возрасте (энцефалит, менингоэнцефалит), эпилепсия в постнатальном периоде.
Симптомы
Фото: kp.ru
Олигофрения является расстройством вследствие органического поражения головного мозга, т.е. связана с нарушением его структуры.
Одним из самых явных симптомов олигофрении является нарушение высших проявлений интеллекта (например абстрактного мышления). Это проявляется в невозможности обучения ребёнка в общеобразовательной школе. В педагогической практике, говоря о таких детях, используют термин задержка психического развития (ЗПР). Также из-за невнимательности, неспособности к запоминанию дети с данным расстройством плохо обучаются даже элементарным вещам, в том числе и уходу за собой. Пациент с олигофренией не способен понимать скрытый смысл происходящего, зачастую не понимает собеседника. Человек с данными расстройствами пытается выйти из неудобного для него положения пустыми размышлениями (при лёгких формах олигофрении).
В эмоциональной сфере также отмечаются расстройства. Пациент не способен понимать и воспроизводить высшие эмоции (стыд, сочувствие, совесть, нравственность, сопереживание, ответственность). Эмоции у таких людей являются простыми и непосредственными, поэтому ,будучи даже в зрелом возрасте, эмоционально они похожи на детей.
Характерно затруднение приобретения элементарных навыков, особенно при тяжёлых формах олигофрении. Это связано не только с неспособностью запомнить и понять действия наставника, но и с невозможностью воспроизведения координированных произвольных движений.
Степень проявления этих симптомов зависит от степени тяжести расстройства, которых выделяют три: идиотия, имбецильность, дебильность.
Диагностика
Фото: proaist.ru
Советским психиатром Сухаревой Г. Е. в 1965 г. были выдвинуты критерии диагностики олигофрении:
- Своеобразная психопатологическая структура слабоумия с преобладанием слабости абстрактного мышления при менее выраженных нарушениях. предпосылок интеллекта и относительно менее грубом недоразвитии эмоциональной сферы.
- Непроцессуальный непрогредиентный характер интеллектуального дефекта
- Замедленный темп психического развития индивида, нарушение онтогенеза (развития организма, начиная от оплодотворения яйцеклетки).
- Необратимый характер расстройств.
При тяжёлых формах олигофрении также могут быть выявлены органические поражения при обследовании пациентов с использованием рентгеновского компьютерного томографа (РКТ), электроэнцефалограммы (ЭЭГ), магнитного резонансного томографа (МРТ): дополнительные полости в головном мозге, большой объём системы желудочков мозга, нарушение соотношения белого и серого вещества, микроцефалия. При сильных органических поражениях возможно изменения формы черепа.
Лечение
Фото: narkolog-psihiatr.ru
Лечение лекарственными средствами взрослых пациентов не позволяет улучшить интеллект. Но большую роль играют выявление и предупреждение олигофрении в младенчестве. Раннее выявление инфекций (сифилиса, листериоза, токсоплазмоза) позволяет начать лечение при отсутствии органических поражений головного мозга. При выявленных энзимопатиях (недостаточность ферментов) назначают специальные диеты (при фенилкетонурии исключают продукты с содержанием фенилаланина), что способствует предупреждению развития расстройств интеллекта. При раннем выявлении олигофрении назначаются препараты, которые могут смягчить последствия расстройств (об этом подробнее в разделе “Лекарства”).
Раннее выявление инфекций (сифилиса, листериоза, токсоплазмоза) позволяет начать лечение при отсутствии органических поражений головного мозга. При выявленных энзимопатиях (недостаточность ферментов) назначают специальные диеты (при фенилкетонурии исключают продукты с содержанием фенилаланина), что способствует предупреждению развития расстройств интеллекта. При раннем выявлении олигофрении назначаются препараты, которые могут смягчить последствия расстройств (об этом подробнее в разделе “Лекарства”).
Важным аспектом в терапии олигофренов с лёгкой степенью поражения интеллекта является социальная терапия. Такие пациенты способны к подражанию. Необходимо трудоустраивать олигофренов в учреждения, где за ними будет осуществляться постоянный контроль со стороны наставника, иначе в силу своей тенденции к отвлечению пациент быстро перестаёт работать.
Лекарства
Фото: popmech.ru
При раннем обнаружении расстройств интеллекта назначают неспецифические метаболические средства, которые способствуют восстановлению, развитию и укреплению центральной нервной системы. Применяются следующие препараты:
- Ноотропы (пирацетам, пиридитол, семакс, фенибут)
- Витамины группы В
- Аминокислоты и их предшественники (глутаминовая и янтарная кислота, церебролизин)
У людей с расстройствами интеллекта выявляется тенденция к беспокойствам, нарушениям поведения и сна. В силу высокой чувствительности мозга с органическими поражениями к нейролептикам стремятся назначать препараты с минимальными побочными эффектами (неулептил, сонапакс, терален, хлорпротиксен). Однако при излишней буйности пациента приходится назначать более сильные препараты (аминазин, галоперидол).
Народные средства
Фото: venskayadacha.com
Народная медицина не в силе восстановить интеллект пациента
Однако можно усмирить страдающего расстройствами интеллекта человека при вспышках буйности и агрессии.
Предлагается следующий сбор трав: приготовить по одной столовой ложке руты душистой и корней валерианы, залить этот сбор кипятком, дать настояться 30 минут. Давать пить человеку в течение дня, перед употреблением процедить.
Давать пить человеку в течение дня, перед употреблением процедить.
При бессоннице принимают ванную с такими травами как календула, мята, душица. Также для улучшения сна прибегают к отварам корня валерианы (2 столовых ложки корня валерианы заливают 1,5 стакана кипятка, 15 минут необходимо нагревать на водяной бане. Принимать после еды 3 столовые ложки перед сном).
Информация носит справочный характер и не является руководством к действию. Не занимайтесь самолечением. При первых симптомах заболевания обратитесь к врачу.Источники
- Руководство по психиатрии. В 2 томах. Т.1/А. С. Тиганов, Р 84 А. В. Снежневский, Д. Д. Орловская и др.; Под ред. А. С. Тиганова. — М.: Медицина, 1999. — 712 с: ил.,[2] л. ISBN 5-225-02676-1
- Н. Н. Иванец, Ю. Г. Ткшышн, В.В. Чирко, М. А. Кинкулышна: Психиатрия и наркология. — М. : ГЭОТАР-Медиа, 2006.-832 с.
- В. П. Самохвалов: Психиатрия. – учебник для медицинских вузов.: 2002 г.- 324 с.
- Основы психиатрии: наглядные материалы к курсу лекций для
0-75 студентов, обучающихся по специальности 1-23 01 04 «Психология» /
сост. А. И. Горбачев. – Минск : БГУ, 2014. – 73 с.
Ваши комментарии о симптомах и лечении
Умственная отсталость по наследству. Риск передачи заболеваний по наследству
всего 21 238 консультаций
Риск передачи сахарного диабета по наследству зависит от вида диабета и степени родства больного человека и ребенка. Даже если риск развития диабета существует – не надо паниковать: оценка факторов риска и коррекция поведения убережет ребенка от болезни.
ВНИМАНИЕ! Перед тем как задать вопрос, рекомендуем Вам ознакомиться с содержанием раздела «Часто задаваемые вопросы». Велика вероятность, что там Вы найдете ответ на свой вопрос прямо сейчас, не тратя время на ожидание ответа от врача – консультанта.Спрашивает Галина:
Может ли по наследству передаться болезнь Альцгеймера?
18 февраля 2013 года
информация о консультанте
Добрый день, Галина!
Болезнь Альцгеймера (деменция) – это заболевание, которое характеризуется постепенной утратой функциональной активности головного мозга, снижением умственных способностей (память, речь, внимание, дезориентация, эмоциональная угнетенность и т. д.). Такая прогрессирующая деменция личности может очень скоро приводить к полной потери дееспособности и в конечном итоге к смерти (кстати, данное заболевание занимает 6 место, среди болезней, приводящих к смерти).
д.). Такая прогрессирующая деменция личности может очень скоро приводить к полной потери дееспособности и в конечном итоге к смерти (кстати, данное заболевание занимает 6 место, среди болезней, приводящих к смерти).
Морфологический субстрат патологии следующий – нервные клетки перерождаются в специфические клубки, и при этом в ткани головного мозга накапливается протеин, образуя так называемые бляшки. Почему именно происходят данные процессы достоверно пока не известно. Надо сказать, что болезнь Альцгеймера чаще всего развивается у людей пожилого возраста (не путать с процессом естественного старения).
А теперь о роли генетического фактора в развитии болезни. Наличие отягощенного семейного анамнеза (наличие родственников, страдающих синдромом Альцгеймера), безусловно, способствует развитию данной патологии. Другими словами можно сказать, что болезнь Альцгеймера – это наследственное заболевание, но проявиться она или нет, будет зависеть от наличия иных провокационных моментов (плохая экология, психо-эмоциональные перегрузки, вредные привычки и т.д.).
Всего доброго!
Спрашивает Алена:
У мужа сестра с задержкой умственного развития. Это наследственная болезнь? Стоит ли бояться, что наш ребенок унаследует это?
19 февраля 2013 года
информация о консультанте
Добрый день!
Умственную отсталость (олигофрения) классифицируют по нескольким категориям (типу, степени, виду и т.д.). Таким образом, различают врожденную и приобретенную олигофрению.
Приобретенное слабоумие характеризуется снижением умственных способностей от нормального уровня, в отличии от врожденного, когда интеллект так и не достигает среднестатистического своего развития.
Сейчас пойдет речь о врожденной умственной отсталости. Итак, среди причин ее возникновения выделяют и наследственную предрасположенность. Считается, что половина случаев умственной отсталости связана как раз с генетическими нарушениями (синдром Дауна, Вильямса, Ангельмана, Ретта и т. д.). Причем степень риска возникновения таковой различна и зависит от каждого конкретного заболевания. И при планировании пополнения в семье обязательно проконсультируйтесь с генетиком и выясните, с чем связано снижение интеллекта у сестры мужа.
д.). Причем степень риска возникновения таковой различна и зависит от каждого конкретного заболевания. И при планировании пополнения в семье обязательно проконсультируйтесь с генетиком и выясните, с чем связано снижение интеллекта у сестры мужа.
Только очная консультация специалиста поможет сориентировать о степени риска в Вашем индивидуальном случае.
Кроме того, нельзя не упомянуть и о влиянии следующих негативных факторов на развитие умственной отсталости ребенка: внутриутробное поражение плода вирусами и бактериями, ионизирующей радиацией, родовая травма, асфиксия в родах и недоношенность.
Всего доброго!
Спрашивает Юлия:
Здраствуйте, подскажите пожалуйста, у родного дяди моего мужа был синдром Дауна, может ли болезнь передаться по наследству ребенку?я очень хочу ребенка, но в тоже время очень боюсь! спасибо большое!! (мне 25 лет, мужу 30 лет).
31 января 2010 года
информация о консультанте
Здравствуйте, Юлия! Синдром Дауна – это генетическое заболевание, причиной которого является наличие в генотипе человека 47 хромосом (вместо 46 в норме), с тремя 21-ми хромосомами. Заболевание возникает в тех случаях, когда происходит неправильное деление половых клеток у родителей, в результате чего образуются гаметы (яйцеклетки или сперматозоиды), содержащие 24 хромосомы (в норме половые клетки содержат 23 хромосомы). Во время зачатия происходит объединение 23-хромосомной клетки одного родителя и 24-хромосомной клетки другого – так получается ребенок с 47 хромосомами (23+24) и синдромом Дауна. К нарушению процесса деления половых клеток приводят многие патологические состояния, в том числе воздействие вредных факторов окружающей среды и зрелый возраст. Риск рождения ребенка с синдромом Дауна увеличивается у женщин после 36 лет, у мужчин – после 40 лет. Из поколения в поколение это заболевание не передается. Ребенок с синдромом Дауна может родиться даже у абсолютно здоровой пары. Для раннего выявления плода с синдромом Дауна проводятся скрининговые обследования беременных на сроке 10-16 недель (УЗИ, маркеры), которые помогают вовремя диагностировать патологию и решить вопрос о целесообразности сохранения беременности. Берегите здоровье!
Берегите здоровье!
Спрашивает Алёна:
Добрый день! Мой муж болеет сахарным диабетом 1-го типа 14 лет, мы подумываем о ребёнке. Куда можно обратиться и какие анализы сдавать, чтобы определить вероятность заболевания сахарным диабетом нашего будущего ребёнка? Я страдаю гипотонией. И возможно ли во время моего рациона питания какими-либо продуктами предупредить заболевание будущего малыша?Заранее благодарна за совет!
23 сентября 2010 года
информация о консультанте
Добрый день, Алена! Во-первых, Вы должны знать о том, что по наследству передается не сам диабет, а лишь определенная склонность (предрасположенность) к нему. А предрасположенность, не равнозначна самому заболеванию. То есть, имея предрасположенность, не обязательно заболеешь самой болезнью. Во-вторых, при диабете I типа (инсулинозависимом) наследственная предрасположенность выражена намного меньше, чем при диабете II типа (инсулиннезависимом). В-третьих, так как диабет есть только у Вашего мужа, то вероятность того, что ребенок в течение жизни тоже заболеет диабетом I типа, не превышает 5%. Другими словами, только 5 из 100 возможных Ваших детей могли бы заболеть диабетом, то есть риск заболеть сахарным диабетом для каждого ребенка весьма невысок. Вы, конечно же, можете посетить генетическую консультацию, но должны понимать, что невозможно при обследовании Вашего мужа, Вас и даже Вашего ребенка выявить тот ген, который предскажет риск диабета. Ведь как говорилось Выше, диабет имеет полигенное (а не моногенное) наследование. К тому же, знание рисков на сегодня все равно не позволяет предотвратить их (рисков) реализацию. Так что, если хотите малыша, беременейте и рожайте. Даст бог, все будет нормально. Будьте здоровы!
Спрашивает Вера:
Скажите пожалуйста! Какова вероятность рождения ребенка с сахарным диабетом или гемофилией,если мой отец болел гемофилией,а младший брат мужа болен диабетом? И может ли генетик запретить рожать,если будет риск этих заболеваний у ребенка?
22 октября 2013 года
Врач эндокринолог высшей категории
информация о консультанте
При планировании беременности надо принимать во внимание и отдаленные последствия – возможность наследственной передачи сахарного диабета от родителей ребенку. В зависимости от типа диабета (1-го или 2-го) различна опасность возникновения у ребенка в будущем сахарного диабета. Вероятность рождения у больной диабетом 1-го типа ребенка, который позже тоже заболеет диабетом, в значительной степени зависит от отца ребенка. Если отец здоров, то вероятность, что ребенок до наступления 20 лет тоже заболеет диабетом 1-го типа — равна всего 1 %.
В зависимости от типа диабета (1-го или 2-го) различна опасность возникновения у ребенка в будущем сахарного диабета. Вероятность рождения у больной диабетом 1-го типа ребенка, который позже тоже заболеет диабетом, в значительной степени зависит от отца ребенка. Если отец здоров, то вероятность, что ребенок до наступления 20 лет тоже заболеет диабетом 1-го типа — равна всего 1 %.
Если диабет 1-го типа имеется у отца, то риск развития в будущем этого диабета у ребенка достигает 6%.
Если у женщины сахарный диабет 2-го типа, то риск для ребенка заболеть диабетом 2- го типа составляет 15 — 30%, если же больны оба родителя, то 60 -70%. Однако сахарный диабет 2-го типа чаще всего развивается после 45 — 50 лет, когда вопрос о беременности уже не так важен для подавляющего большинства женщин. Запретить беременность генетик не может. Но обследоваться надо и врач даст советы по контролю, питанию во время беременности.
С уважением, Наталья Васильевна.
Спрашивает Оля:
Скажите пожалуйста если у моего будущего мужа врожденная астма, которая передалась ему от его отца на 50%, то его ребенок так же родится с врожденной астмой или есть все таки шансы что он будет здоров?
16 сентября 2013 года
информация о консультанте
Добрый день. Бронхиальная астма, как заболевание, по наследству не передается. Но передается предрасположенность к развитию аллергических заболеваний, в том числе и бронхиальной астмы. Вероятность передачи по наследству этой предрасположенности составляет 20-30%. Таким образом, в 70-80% рождается здоровый ребенок, без склонности к аллергии. Следует учитывать, что большое значение имеют пусковые факторы в развитии астмы. Поэтому если ребенок унаследует предрасположенность к аллергическим заболеваниям, то развитие астмы можно предотвратить, соблюдая определенные условия. Начинать нужно с подготовки к беременности, вести здоровый образ жизни, соблюдать гипоаллергенную диету и гипоаллергенный быт во время беременности, грудного вскармливания, первых лет жизни ребенка. С первых дней жизни малыша стоит заниматься неспецифической профилактикой инфекционных заболеваний и их осложнений. Более подробную информацию о наследственных заболеваниях можно получить при медико-генетическом консультировании у генетика. Всего доброго.
С первых дней жизни малыша стоит заниматься неспецифической профилактикой инфекционных заболеваний и их осложнений. Более подробную информацию о наследственных заболеваниях можно получить при медико-генетическом консультировании у генетика. Всего доброго.
Спрашивает Николай:
Добрый день! Может ли по наследству передаться болезнь Паркинсона? Спасибо.
18 февраля 2013 года
информация о консультанте
Здравствуйте, Николай!
Болезнь Паркинсона — это заболевание характеризующееся поражением определенных структур головного мозга (substantia nigra), что приводит к уменьшению выработки такого биологически активного вещества как дофамин (который участвует в передаче нервных импульсов). Клинически это проявляется скованностью движений, тремором, забывчивостью, изменением характера и т.д.
А теперь по сути вопроса. Данное заболевание носит наследственный характер и наследуется по аутосомно-доминантному типу, но неполной пенетрацией, это значит, что хотя ген доминирует, но проявиться может не всегда. Поэтому тут более уместным будет говорить о передачи по наследству склонности к развитию болезни Паркинсона.
Чаще всего заболевание развивается после травмы, сильного психо-эмоционального перенапряжения, а также у людей старшего и пожилого возраста. Поэтому профилактика и своевременное лечение смогут помочь предотвратить как появления самого заболевания, так и облегчить его течение. Всего доброго!
Спрашивает Ирина:
Здравствуйте. Подскажите пожалуйста,если у меня был обнаружен цитамегаловирус герпеса,может ли это повлиять на здоровье будущего ребенка.?
25 июля 2013 года
информация о консультанте
Добрый день. Цитомегаловирусная инфекция – довольно распространенное инфекционное заболевание. При выявлении цитомегаловируса важным моментом является, как именно его обнаружили. Обычно определяют иммуноглобулины М, G и ДНК вируса (с помощью ПЦР). Иммуноглобулин М указывает на острое заболевание, при определении ДНК вируса можно определить активность процесса. Иммуноглобулины G указывают на давнее заражение цитомегаловирусом, но не характеризуют активность процесса. Для плода и новорожденного опасны острый процесс и активация хронической инфекции во время беременности. В этом случае развивается врожденная цитомегаловирусная инфекция, которая приводит к гибели плода или нарушению его развития, при рождении таких детей у них отмечается разнообразная клиническая картина, но наиболее часто умственная отсталость и нарушения слуха. Для того чтобы предупредить такие проблемы необходимо планирование беременности, обследование на инфекции до беременности и своевременное лечение. Всего доброго.
Иммуноглобулины G указывают на давнее заражение цитомегаловирусом, но не характеризуют активность процесса. Для плода и новорожденного опасны острый процесс и активация хронической инфекции во время беременности. В этом случае развивается врожденная цитомегаловирусная инфекция, которая приводит к гибели плода или нарушению его развития, при рождении таких детей у них отмечается разнообразная клиническая картина, но наиболее часто умственная отсталость и нарушения слуха. Для того чтобы предупредить такие проблемы необходимо планирование беременности, обследование на инфекции до беременности и своевременное лечение. Всего доброго.
Спрашивает Марина:
Мне 26 лет. С детства болею бронхиальной астмой и аллергией. Проблемы появились после прививки. Раньше заболевания проявлялись в очень тяжелой форме, сейчас все заметно лучше. В моей семье и семье моего мужа не болеют.Могу ли я родить здоровых детей? Что для этого нужно делать? Как не передать мои заболевания детям и внукам?
12 августа 2013 года
информация о консультанте
Добрый день. Риск передачи бронхиальной астмы по наследству составляет 20-30%. Аллергические заболевания не являются наследственными, однако по наследству передается предрасположенность к аллергии. В возникновении этих заболеваний играет роль не только наследственность, а и много других провоцирующих факторов. Чтобы предупредить развитие аллергических заболеваний и бронхиальной астмы у ребенка необходимо заботится о здоровом образе жизни ребенка с первых дней. Начинать нужно с грудного вскармливания и гипоаллергенного питания матери во время лактации. Создание гипоаллергенного быта заключается в избавлении от пыли, частой влажной уборке, ограниченном использовании химических средств для дома и личной гигиены, проветривании, особом подходе к домашним животным. Закаливание и неспецифическая профилактика инфекционных заболеваний укрепит иммунитет ребенка. Более подробные рекомендации можно получить у детского аллерголога и медико-генетической консультации. Всего доброго.
Всего доброго.
Спрашивает Наталья:
Здравствуйте! У моего мужа от первого брака 2 детей. Младшая дочь болеет диабетом 1 типа с пяти лет. Сейчас ей 11. Старший брат,родители и близкие родственники девочки не болеют диабетом. У меня папа и бабушка по линии отца, диабет 2 типа. У меня самой и моей 15 летней дочери сахар в норме. Какова вероятность заболевания у моего будущего ребенка? Заранее спасибо.
12 июля 2013 года
информация о консультанте
Добрый день.В развитии сахарного диабета наследственность имеет значение. Диабет первого типа передается по наследству в 3-7% случаев. Диабет второго типа передается значительно чаще, что составляет около 50% случаев. Однако при отсутствии наследственных факторов тоже есть риск возникновения этого заболевания (около 0,25%). Поэтому сказать наверняка, что у Вашего мужа, а соответственно и Вашего ребенка, есть предрасположенность к сахарному диабету первого типа нельзя. А вот предрасположенность к сахарному диабету второго типа будет. При этом следует помнить, что если соблюдать меры профилактики, то вероятность развития сахарного диабета 2 типа значительно падает. Более подробно о рисках развития диабета можно узнать на нашем портале: Диабет второго типа, или последствия «сладкой жизни» Всего доброго.
Спрашивает Игорь:
Насколько опасен Псориаз у матери для будующего ребёнка?
10 сентября 2009 года
информация о консультанте
Добрый день! Поскольку псориаз, согласно одной из теорий, заболевание наследственное — Ваш вопрос, Игорь, совершенно правомочен. Генетики считают, что при наличии псориаза у одного из родителей вероятность появления этого заболевания у ребенка увеличивается на 25%. А вот дерматологи утверждают, что наследуется не само заболевание, а особые свойства организма (строение кожи, особенности работы системы иммунитета). Запускает же развитие псориаза воздействие на такой организм факторов риска. К факторам риска относятся нервные и физические перегрузки, наличие очагов хронической инфекции (гайморит, кариес, пиелонефрит, аднексит и т. д.), переедание и злоупотребление сладостями, чрезмерное пребывание на солнце, употребление ряда лекарственных препаратов (антибиотиков, ферментных препаратов). Поэтому, если оберегать ребенка с предрасположенностью к псориазу от воздействия провоцирующих факторов, вероятность развития болезни можно сильно уменьшить. Будьте здоровы!
д.), переедание и злоупотребление сладостями, чрезмерное пребывание на солнце, употребление ряда лекарственных препаратов (антибиотиков, ферментных препаратов). Поэтому, если оберегать ребенка с предрасположенностью к псориазу от воздействия провоцирующих факторов, вероятность развития болезни можно сильно уменьшить. Будьте здоровы!
Спрашивает Яна:
Добрый день!подскажите пожалуйста какой у нас шанс родить здорового ребенка,если у мужа атопический дерматит,пере кручен желчный пузырь,астма,песок в почках,очень плохие зубы,практически нет иммунитета?
26 апреля 2013 года
информация о консультанте
К сожалению, при наличии таких аллергических заболеваний как атопический дерматит и бронхиальная астма, риск развития, в течение, жизни у ребенка данных состояний достигает 30-40%. Но, следует отметить, что по наследству передаются не заболевания, как таковые, а предрасположенность к ним. Поэтому, используя все доступные профилактические меры: как можно более длительное грудное вскармливание, коррекция микрофлоры кишечника и мамы и ребенка, избегание потенциальных аллергенов как во время беременности так и ребенком в дальнейшем (а именно: коровье молоко, шоколад, орехи, мед,рыба, цитрусовые), использование гипоаллергенных предметов бытовой химии, установка систем доочистки воды, употребление рыбьего жира, как источника омега-3 кислот позволить как можно больше снизить риск развития у ребенка данных состояний. Еще раз следует подчеркнуть важность сохранения грудного вскармливания у ребенка предрасположенного к аллергическим заболеваниям.
Спрашивает Елена:
Муж болен раком,,были лучи.Можно ли рожать ребёнка?
20 августа 2013 года
информация о консультанте
Добрый день. После лучевой терапии отмечается влияние на репродуктивную функцию мужчин. Обычно после этой процедуры наблюдается снижение количества сперматозоидов, снижается их подвижность. Это приводит к бесплодию мужчин, которое обычно носит временный характер, постепенно репродуктивная функция восстанавливается. При сохраненной функции возможно развитие генетических нарушений в структуре сперматозоида, которое повлечет генетическое нарушение у потомства. Однако в настоящее время нет точных данных, в каких случаях появляется вероятность развития генетических нарушений и каких именно генетических нарушений, что тоже имеет большое значение. Если беременность уже наступила, то лучше обратиться для медико-генетического консультирования, а также внимательно вести свою беременность, проходить все необходимые обследования в положенный срок, чтобы на ранних этапах можно было выявить возможные отклонения. Всего доброго.
Спрашивает Александр:
Здравствуйте у меня такой вопрос.У моего сына обнаружили врождённый порок сердца ,из-за чего он может быть если не у моей семьи и семьи моей жены не было проблем с сердцем ни у кого .Отправляют на операцию в Москву !
11 июня 2013 года
информация о консультанте
Добрый день. Риск передачи заболеваний сердца по наследству составляет всего 5%. Основная причина развития врожденных пороков сердца – это влияние неблагоприятных факторов на будущую мать во время беременности. Формирование сердца у плода начинается с 4 недели внутриутробного развития, самый важный период с 6 по 8 неделю. Это время, когда некоторые женщины еще не знают о своей беременности и подвергают опасности будущего ребенка. Вызывать развитие пороков сердца могут вирусные заболевания, некоторые лекарства, неблагоприятные излучения, стрессы, алкоголь, курение, наркотики. Более подробно о пороках сердца и причинах их возникновения можно узнать на нашем портале в статье: Врожденный порок сердца – что стоит за диагнозом Всего доброго.Спрашивает Екатерина:
Добрый день. У моей матери стоит диагноз — олигофрения. При моём рождении врачи сказали, что вероятность передачи равна 50 на 50. У меня вроде никаких отклонений нет, хотя есть некоторые трудности с общением с людьми, но это скорей родом из детства и не принимания меня в школе.
Какова вероятность того, что это передастся моим детям?
Внукам?
С уважением, Екатерина.
10 октября 2013 года
информация о консультанте
Здравствуйте, Екатерина! Судя по стилю и орфографии Вашего сообщения, особых проблем с умственным развитием у Вас нет. Нарушения, передающиеся по наследству, — это лишь одна, небольшая группа причин, вызывающих олигофрению. Довольно часто в развития олигофрении лежат токсическое или инфекционное воздействие на плод во время беременности, травма или неправильное питание/голодание беременной женщины, тяжелая несовместимость матери и плода по резус-фактору или группе крови, травматичные роды (родовая травма), эндокринные заболевания, травмы, инфекции или токсические воздействия на ребенка в первые годы жизни. В этих случаях интеллектуальный дефект, присутствующий у одного члена семьи, по наследству не передается. Если есть сомнения — можно пройти обследование у генетика и психиатра — врачи с пониманием отнесутся к Вашим опасениям и обязательно ответят на все Ваши вопросы. Берегите здоровье!
всего 10 страниц
Абсолют-Помощь. Благотворительный фонд.
Что такое умственная отсталость?
Умственную отсталость у детей относят к недоразвитости всех процессов психического развития, которая проявляется в низком уровне интеллекта, когда ребенок не способен познавать окружающий мир, обучаться, приобретать знания в соответствии со своим биологическим возрастом. Согласно врожденным или приобретенным факторам такие дети могут развиваться до определенного лимита своих возможностей. В медицинском справочнике заболевание носит название «олигофрения». Чем скорее родители ребенка с диагнозом умственной отсталости примут его индивидуальные особенности, тем быстрее они смогут начать корректирующие и поддерживающие мероприятия.
Заторможенное восприятие отдельных предметов требует не только больше времени, но и не позволяет проводить между ними соответствующую взаимосвязь. Графические предметы, буквы, цифры, похожие по звучанию слова у детей с данным диагнозом помимо замедленного и ограниченного восприятия, путаются, смешиваются. Ребенок не может воспринимать важную информацию, выхватывая лишь фрагменты. Он не может дать анализ или описание картинке или предмету. Обратив внимание на определенный предмет, ребенок не переходит к изучению следующего, поэтому нуждается, чтобы его побуждали к действиям.
Основное отличие от здоровых детей заключается в сложности воспроизведения полученной информации из-за нарушенной мыслительной деятельности и дефекта речи. Овладение чтением и письмом является довольно сложным процессом. Дети с этим заболеванием невнимательны, не способны доводить начатое до конца. Недоразвитость отображается и на эмоциональной сфере, когда эмоции отсутствуют или ограничены, а различные переживания не проявляются. Нарушены волевые процессы, дети не активны. Они позже начинают держать головку, ползать, ходить, проявлять интерес к предметам и различать их, лепетать, узнавать родных или чужих людей.
Согласно Международной классификации болезней выделяют следующие виды умственной отсталости:
- Легкая умственная отсталость. Внешне такие люди могут совсем не отличаться от здоровых людей. Обычно они испытывают сложности в обучении из-за сниженной способности к концентрации внимания. При этом память у них достаточно хорошая. Подавляющее большинство из них достигают также полной независимости в сфере ухода за собой и в практических и домашних навыках, даже если развитие происходит значительно медленнее, чем в норме. Основные затруднения обычно наблюдаются в сфере школьной успеваемости. Тем не менее, при легкой умственной отсталости значительную помощь может принести образование, предназначенное для развития их навыков и проявления компенсаторных возможностей. В большинстве благоприятных случаев легкой умственной отсталости возможно трудоустройство, требующее способностей не столько к абстрактному мышлению, сколько к практической деятельности. Опыт показывает, что у детей, переживших травму потери кровных родителей, воспитывающихся в сиротских учреждениях, подвергавшихся насилию, жестокому обращению, зачастую трудно различить симптомы умственной отсталости и социопедагогической запущенности. При изменении условий жизни на более благоприятные эти дети, как правило, дают хорошую динамику в развитии и их диагноз может быть снят или пересмотрен.
- Умеренная умственная отсталость. Люди с таким диагнозом способны испытывать привязанности, различать похвалу и наказание, их можно обучить элементарным навыкам самообслуживания, а иногда и чтению, письму, простейшему счету. Образовательные программы могут дать им возможности для развития своего ограниченного потенциала и приобретения некоторых базисных навыков. В зрелом возрасте лица с умеренной умственной отсталостью обычно способны к простой практической работе при тщательном построении заданий и обеспечении квалифицированного сопровождения. Совершенно независимое проживание достигается редко. Тем не менее, такие люди в общем полностью мобильны и физически активны и большинство из них обнаруживают признаки социального развития, что заключается в способности к установлению контактов, общению с другими людьми и участию в элементарных социальных занятиях.
- Тяжелая умственная отсталость. Люди с таким диагнозом могут частично овладеть речью и усвоить элементарные навыки для обслуживания себя самого. Словарный запас очень скудный, порой он не превышает десяти-двадцати слов, мышление очень конкретное, хаотическое и бессистемное. Почти у всех детей этой категории заметны большие двигательные нарушения.
- Глубокая умственная отсталость. У людей с глубоким поражением мозга, очень часто нарушены строения внутренних органов. Их речь не развивается, она полностью нарушена. С самого раннего детства заметно их отставание в развитии, малыши поздно начинают сидеть и держать головку. При тяжелой форме умственной отсталости дети малоподвижны. При таком диагнозе на первое место выходит медицинский уход за ребенком, услуги социальной няни.
Первичные проявления умственной недостаточности у детей обычно включают в себя такие симптомы и признаки как отставание интеллекта, инфантильное поведение, недостаточные навыки по самообслуживанию. Такое отставание становится очень заметным к дошкольному возрасту. Однако при лёгкой умственной отсталости такие симптомы могут не проявиться до школьного возраста. Намного раньше отсталость интеллекта диагностируется при наличии умеренной и тяжёлой степени этого нарушения, а также тогда, когда отсталость умственного развития сочетается с дефектами развития и физическими пороками. Среди детей дошкольного возраста явным признаком является наличие сниженного уровня IQ в сочетании с ограниченностью проявления адаптивных навыков поведения. Хотя индивидуальные особенности этого нарушения могут меняться, чаще у детей с недостаточностью интеллекта отмечается скорее постепенный прогресс, а не полная остановка развития.
Важно помнить, что чем раньше начать реабилитацию, тем лучше. Также первостепенная задача – ранее выявление заболевания. Однако проблема заключается в том, что очень сложно диагностировать умственную отсталость на ранних сроках развития ребенка. Как правило, подозрения у родителей возникают с двух лет, когда ребенок не говорит или говорит плохо. И только ближе к трем-четырем годам ставят диагноз «умственная отсталость», поскольку проблема становится явной.
Родителям следует обратить внимание:
- насколько хорошо и уверенно ребенок держит головку;
- когда он научился сидеть самостоятельно;
- как ребенок ползает;
- как произносит первые звуки, лепечет.
Внимание! Если есть подозрения на отставание ребенка в интеллектуальном развитии, следует немедленно обратиться к специалистам.
Признаки умственной отсталости у детей до 1 года
Дети с умственной отсталостью в возрасте до 1 года выявляются при осмотре врачами, опираясь на отставание в психомоторном развитии или на обнаружение признаков наследственных (хромосомных и генетических) синдромов. Родители часто лишь могут указывать на то, что ребенок так и не начинает концентрировать взгляд, плохо растет, не пытается разговаривать и так далее
Основные и косвенные признаки умственной отсталости у детей до года:
- Характерные морфологические признаки наследственных синдромов (эпикант глаз, одна борозда на ладони при синдроме Дауна; «лицо эльфа», пороки сердца и крупных магистральных сосудов при синдроме Вильямса; характерные складки шеи при синдроме Шерешевского-Тернера и другие).
- Психическая заторможенность – неадекватная реакция на кормление, спонтанная (без видимой причины) перемена эмоций.
- Отсутствие «контакта глаз» и следящего движения ими по достижении младенцем возраста старше 4 месяцев.
- Сохранность врожденных рефлексов и их спонтанное возникновение.
- Судорожные припадки.
- Отсутствие попыток ползать и садиться.
- Отсутствие у малыша попыток звукоподражания взрослым («гуления»).
- Явления самоповреждения.
К сожалению, получить полную картину нарушения когнитивных функций у ребенка с признаками в замедлении развития, в том числе и умственного, сложно ввиду возраста, несформировавшейся речи и ограничения других коммуникативных возможностей.
Поэтому диагностика такой патологии требует комплексного подхода и проведения следующих обследований и консультаций:
- Осмотр неонатолога или педиатра, установление признаков наследственных синдромов или общего отставания в развитии.
- Консультация детского невролога. Оценка функционального состояния ЦНС, проведение МРТ головного мозга для выявления органической патологии.
- Генетические анализы и оценка кариотипа для подтверждения диагноза наследственных синдромов у новорожденного.
- Консультация детского инфекциониста в случае подозрения инфекционной причины интеллектуального отставания.
- Консультация гематолога при тяжелом течении гемолитической болезни.
Далее такие дети наблюдаются врачом-педиатром по диспансерному плану (с определенной регулярностью) с целью оценить степень умственной отсталости, своевременно решить вопрос о присвоении группы инвалидности и назначении специализированного лечения (при необходимости). К сожалению, в таком возрасте довольно сложно определить точный прогноз относительно степени тяжести умственной недостаточности, которая сформируется у ребенка в будущем.
Признаки интеллектуального отставания у детей после 1 года
У маленьких пациентов, которые обследуются уже в возрасте старше 1 года, установление точной причины и степени когнитивных нарушений гораздо проще. Также в дошкольном периоде выявляются нетяжелые формы умственной отсталости, которые заключаются в неспособности к синтезу и анализу полученной информации, ассоциативному и логическому мышлению. Таким образом, признаками умственной недостаточности у детей являются:
- Нарушение речевого развития и способности к коммуникации со взрослыми и сверстниками. Довольно бедный словарный запас. Зачастую такие дети с трудом обучаются письму и чтению.
- Несдержанное, порой агрессивное поведение, возникающее спонтанно или как неадекватная реакция на окружающий мир.
- Психолого-педагогическая характеристика детей с умственной отсталостью в дошкольный период указывает на их способность с трудом овладевать новой информацией, выявляются нарушения самообслуживания. Почти в 50% случаев имеют место признаки нарушения психики, требующие консультации соответствующих специалистов.
- Характеристика тестов для оценки интеллектуального уровня, способности к логическому и ассоциативному мышлению выявляют наличие различной степени умственной недостаточности.
- Агрессивность к себе или окружающим, несвязанная с явными причинами.
Карта обследования таких детей должна учитывать необходимость не только установления предположительной причины отставания в умственном развитии, но и давать объективную оценку степени прогрессирования патологии. Если установлено, что симптомы отставания в развитии наблюдались еще с момента рождения, врач подробно изучает, как протекала беременность у матери, ход родов и имевшие место заболевания первого года жизни. Обычно наиболее распространенные наследственные синдромы в этом возрасте имеют явную клиническую картину, и нет необходимости проводить ребенку анализ кариотипа (строения и количества хромосом в ядрах клеток).
Иногда явления умственной неполноценности наряду с другими патологиями могут быть признаками редких генетических болезней, которые требуют сложной, часто дорогостоящей диагностики для постановки точного диагноза.Также врачу важно установить, не сопровождались ли респираторные инфекции осложнениями со стороны центрального отдела нервной системы (менингит, энцефалит и так далее). Для этого применяют следующие методики диагностики, помогающие в установлении причины интеллектуального дефицита:
- Изучение истории болезни ребенка, установление наличие тяжелых травм или инфекций.
- МРТ или КТ головного мозга.
- Иммунологические анализы для выявления антител к возбудителям венерических заболеваний, могущих передаться от матери, менингококковой и других инфекций.
- Проведение ЭЭГ для оценки волновой активности коры головного мозга.
- Консультация детского невролога.
Что делать, если ребенку поставили диагноз «умственная отсталость»?
Шаг 1. Обратиться к неврологу, имеющему опыт работы с этим заболеванием (для получения бесплатных услуг в государственных центрах, необходимо взять направление у районного невролога).
Шаг 2. Пройти консультацию психиатра.
Шаг 3. Провести полную диагностику здоровья на предмет сопутствующих заболеваний.
Шаг 4. В случае умеренной и тяжелой умственной отсталости оформить необходимую группу инвалидности и вписать все необходимое в ИПРА (индивидуальный план реабилитации и абилитации)
Шаг 5. Начать реабилитацию, занятия с дефектологом, логопедом, психологом.
О ранней диагностике и комплексной реабилитации
Ресурсы для родителей
Важно начать реабилитацию как можно раньше!
Главная роль в реабилитации детей с умственной отсталостью принадлежит педагогическому процессу. Необходимо обратиться за помощью и поддержкой к квалифицированным специалистам. Ключевой фактор прогресса – контакт родителей с врачами, педагогами, психологами. Родители должны понять, что наилучшая адаптация возможна лишь в том случае, если программа обучения будет соответствовать возможностям ребенка. Залогом успешного лечения умственной отсталости является комплексное воздействие, то есть применение не только медикаментов, но и индивидуальный подход к обучению, занятия со специалистами – все это необходимо для более успешной адаптации ребенка в окружающем мире.
Основные медицинские рекомендации родителям детей с умственной отсталостью выглядят следующим образом:
- Вовремя обратитесь за медицинской помощью и выясните причину и степень прогрессирования умственной неполноценности.
- Регулярно проводите обучающие и развивающие занятия с таким ребенком. Коррекция поведенческих отклонений может быть проведена со специальным детским психологом.
- Старайтесь не изолировать ребенка от сверстников и взрослых, несмотря на все опасения.
- Социальная адаптация детей с умственной отсталостью должна стать основой обучения и воспитания. Лишь достижение самостоятельности в быту, основ речи и письма, а также овладение простыми профессиями позволит во взрослой жизни быть полноценным членом общества и не требовать посторонней помощи.
- Не завышайте планку ожидаемых результатов для таких детей. Часто особенности характера и повышенное чувство собственной неполноценности и переживание неудач сводят на нет все достигнутые результаты. С другой стороны, не нужно потакать лени ребенка или останавливаться на достигнутом якобы приемлемом результате. Это часто проявляется при обучении общению – родители понимают желания ребенка по звукам или искаженным словам, однако для социальной адаптации и коммуникации с другими людьми этого недостаточно.
О лечении:
Куда обратиться
Государственные центры
Центру более 250 лет. Центр структурирован так, что и в научном, и в практическом плане решает мультидисциплинарные вопросы профилактики, диагностики, лечения, высокотехнологичного лечения и реабилитации. В условиях круглосуточного пребывания, дневного стационара и консультативно-диагностического центра оказываются все виды медицинской помощи, включая специализированную и высокотехнологичную. Новые медико-организационные формы работы позволили ежегодно увеличивать число пациентов, прошедших курс лечения, в 1,5 раза. Все виды исследований в Центре являются общедоступными для детей из любых субъектов РФ.
Адрес: г. Москва, Ломоносовский проспект, 2, стр.1
Тел.: +7 (495) 967-14-20
Электронная почта: [email protected]
Адрес: г. Санкт-Петербург, ул. Бехтерева, 3
Тел.: +7 (812) 670-02-34
Электронная почта: [email protected]
Негосударственные центры
Создан в 1989 году по инициативе родителей и педагогов для помощи детям с различными особенностями развития. В Центре лечебной педагогики занимаются дети с расстройствами аутистического спектра, эпилепсией, генетическими синдромами, нарушениями умственного развития, трудностями обучения и другими проблемами.
Адрес: г. Москва, ул. Строителей, д. 17Б
Тел.: +7 (499) 131-06-83, 133-84-47, +7 (495) 646-50-66, 930-00-01
Электронная почта: [email protected]
Олигофрены у Вечного огня. Путин навсегда. Кому это надо и к чему приведет?
Олигофрены у Вечного огня
Эта жуткая история произошла перед Новым 2008 г. в городе Кольчугино Владимирской области. Четверо отморозков сожгли заживо на Вечном огне молодого рабочего Алексея Денисова. Казнили за то, что он сделал им замечание — нельзя пить пиво на мемориале. Все в этой страшной истории поражает своей обыденностью. В день своей гибели Алексей вышел «проветриться». Это было в десять вечера. Он обещал скоро вернуться. Родители прождали его всю ночь. Следующий день искали по знакомым. И только на третий день узнали, что на Вечном огне сожгли мужчину, который обгорел настолько, что невозможно опознать. Родные узнали его по свитеру. У него было типичное лицо русского северянина. Курносый нос. Широко расставленные глаза. Тихий парень. Работал на металлургический заводе им. Орджоникидзе. Всю зарплату отдавал семье. Мог выпить, но пьянство не одобрял, потому что был очень религиозен. Занимался спортом. Много читал, любил русские народные и советские песни. Очень гордился прадедом, дошедшим до Берлина. Наверное, поэтому и вступился за мемориал. Корреспондент «Новой газеты» Елена Костюченко выяснила, побывав в Кольчугино, что трое из четверых убийц Алексея — 20-летние Михаил Данилов, Николай Курагин, Александр Андреев — выпускники Кольчугинской коррекционной школы-интерната. Четвертый — 14-летний Алексей Горячев. Директор этого интерната рассказал Елене вот что: «Здесь учатся дети, диагноз которых не позволяет учиться в обычной школе. Обычно этот диагноз — олигофрения. Так, Данилов и Курагин проходили лечение в психиатрической больнице. Олигофрены часто попадают в милицию. У них животные инстинкты преобладают над чувствами. То есть им хочется есть — и они возьмут булочку с прилавка и не задумаются о последствиях. Потом, они очень ведомые. Если ты старше — то есть они воспринимают тебя как взрослого, — и ты предлагаешь им яблоко, шоколадку, они все сделают по твоей просьбе. Если вор попросит их помочь вынести вещи из квартиры — они вынесут. Они не задумываются. Потому что не могут. У меня это убийство из головы не идет. С олигофренами нельзя разговаривать грубо: они немедленно возбуждаются. А он вроде делал им замечания… Да, у неполноценных людей бывают приступы агрессивности чаще, чем у других. Убийство произошло на Вечном огне… А вот я уверен, что они даже не очень соображали, что это за место, где они пили пиво. Для них это был просто огонь, костер, тепло. Вы не видите того, что вижу я. Олигофрения передается по наследству, и в семьях олигофренов пять-семь детей — норма. И дети наших выпускников возвращаются к нам. В некоторых деревнях около Кольчугина живет уже пятое поколение олигофренов. И из-за тотального пьянства в их полку постоянно прибывает. Это — вырождение. Вот по телевизору тут говорили, что во Владимирской области рождаемость превысила смертность впервые за 25 лет. Да, это так. Но говорят только о количественных показателях, не о качественных. Мы отслеживаем судьбу выпускников два-три года. Но это, как правило, бессмысленно. Они, выходя из нашей школы, возвращаются в те же условия, от которых они убегали, — пьяные родители, комнатка в общежитии. И все идет по кругу…» («Новая газета», 14.02.2008.).
Директор Кольчугинского интерната поставил точный диагноз — рост числа олигофренов в стране свидетельствует о вырождении нации. Это неизбежное следствие того, что в России все больше людей спиваются. Дети, которые от них рождаются, как правило, умственно отсталые, неполноценные. Олигофрены при этом очень похотливы. Они не предохраняются, потому что не понимают, зачем это нужно. Так из поколения в поколение в геометрической прогрессии воспроизводятся У. О., что на кодовом языке психиатров означает «умственно отсталые». Им нельзя иметь детей, ибо с появлением у них потомства генофонду нации наносится непоправимый ущерб. Первопричина тому — многолетняя алкоголизация и наркотизация населения России.
Число У. О. на 1000 человек населения в России выше, чем в большинстве стран мира. По данным статистики в Российской Федерации насчитывается 1,7 миллиона детей с ограниченными возможностями, что составляет 4,5 % всей детской популяции. Это выше допустимых норм ВОЗ более чем в два раза. (См. А. Ц. Цыбикова. Система специализированной помощи детям с ограниченными возможностями. Улан-Удэ, 2002) Умственно отсталые дети различаются по степени выраженности дефекта. Дети с легкой степенью У. О. (дебилы) составляют 75–80 %. После окончания специальной (коррекционной) школы, вроде Кольчугинской, многие из них трудоустраиваются и социально адаптируются. Дети со средне выраженной отсталостью (имбецилы) — их 15–20 % — обучаются по специальным программам в специализированных классах, осваивают элементарные навыки письма, чтения, счета, простейшие трудовые операции (склеивание коробок, конвертов), бытовые навыки. Эта категория детей имеет статус инвалида. Дети с глубоко выраженной умственной отсталостью (идиоты) — 5 % — живут и воспитываются в интернатных учреждениях Министерства труда и социального развития, где остаются пожизненно. Данная категория детей также имеет статус инвалида.
Вдумайтесь в этот термин «социально адаптируются». Трое таких «адаптировавшихся» вместе с примкнувшим к ним подростком-алкоголиком и сожгли Алексея на Вечном огне в Кольчугино. Они имели паспорта. Имели право покупать спиртные напитки. И активно этим правом пользовались. Могли получить даже водительские права при определенном напряжении своих параллельных позвоночнику извилин. О неминуемых последствиях этого расскажет любой гаишник. Они могли создать семью, официально зарегистрировавшись в кольчугинском ЗАГСе. У них могли родиться дети, не обязательно, но, скорее всего — такие же олигофрены или дебилы, имбецилы, идиоты… Они по-своему — несчастные существа, потому что не понимают, где они, что творят, зачем. Они не понимали, что убивают, лишают человека жизни. Олигофрены начисто лишены способности к обобщению, абстрактному и математическому мышлению. Они — одноклеточные. Ничего не читают. Только смотрят телевизор, понимая далеко не все и воспринимая все буквально. Они никогда не осознают в силу своей врожденной ущербности, что такое патриотизм, Родина, любовь, дружба…. Хотя их вполне можно научить убивать, грабить, насиловать. Вплоть до конца своих жалких дней они обречены на участь неполноценных и неадекватных существ с человеческим обликом, но без человеческой сущности. Число их в России катастрофически растет. Это заметно, особенно в метро. Достаточно вглядеться в лица, когда едешь на эскалаторе. Все чаще встречаешь кольчугинских двойников — ни единой мысли на неандертальском челе. От этого жутко, страшно. Воочию видишь, что М. Фешбах правильный сделал вывод в своей статье в «Вашингтон пост» — русская нация вымирает…
За последние два десятилетия в РФ более чем на 47 % выросла первичная заболеваемость состоянием слабоумия и психозами. По данным РАМН за 1999 год примерно у 52 миллионов человек (а это треть населения страны) имеются скрытые психические нарушения, по поводу которых люди не обращаются за медицинской помощью. Но это означает, что при различных стрессовых ситуациях, волнениях, переживаниях, связанных с различными духовно-психологическими проблемами, легко могут произойти психический срыв или инициация психического заболевания, а у подростков и молодежи это является и важным стимулом обращения к наркотикам, чтобы уйти от реалий нашей жизни. В некоторых регионах страны (Читинская область) заболеваемость олигофренией превышает в 4,5 раза общероссийские показатели, а заболеваемость олигофренией у подростков в 6,8 раза превышает показатель по России. В Псковской области «каждый третий ребенок рождается больным олигофренией», — сообщила заместитель председателя Псковского общества трезвости и здоровья Людмила Костина. В 1985 году этот жуткий показатель был куда ниже — только каждый пятый ребенок на Псковщине, этой исконно русской земле, рождался олигофреном. Такой «прогресс» наоборот, как пояснила г-жа Костина, — «результат чрезмерного потребления алкоголя: на каждого жителя Псковской области, включая младенцев и стариков, приходится 14 декалитров чистого алкоголя, А 8 дал (декалитров) уже считается смертельной дозой, и, как следствие, дети рождаются олигофренами». Всего же на учете в Псковской области состоит 13 332 У. О., что в относительных цифрах составляет — 1713,6 на 100 тыс. человек населения. Жуткая цифра. Но, как сообщили в областном наркологическим диспансере, эти цифры на протяжении последних лет остаются стабильными, (см. www.newsru.com). И так по всей стране.
Особо трагическая ситуация складывается с более 700 тыс. детей-сирот и детей, оставшихся без попечения родителей. Многих из них сама жизнь в России отбрасывает в категорию У. О. и неполноценных, обрекая на то же и их потомство. Примерно треть из них воспитывается в «казенных» учреждениях — домах ребенка, детских домах, школах-интернатах общего типа, интернатах для детей с ограниченными возможностями здоровья и т. д. По статистике 40 % находившихся на полном государственном обеспечении сирот впоследствии становятся алкоголиками и наркоманами, 10 % кончают жизнь самоубийством, 40 % совершают преступления. В то же время, если большинство стран стремятся развивать альтернативные формы устройства детей, оставшихся без попечения родителей, в России при неуклонном росте общего числа сирот (по данным Госкомстата, только за последние 3 года — на 83 тыс.) практика устройства в семьи и передачи под опеку постепенно сворачивается. В результате ежегодно все больше детей попадает в условия, затрудняющие, а зачастую и исключающие полноценное развитие личности. Все чаще в российских СМИ рассказывают о детях-маугли — брошенных родителями на произвол судьбы и воспитанных бродячими животными.
«Российская система воспитания сирот приписывает детям, воспитывавшимся в государственных учреждениях, склонность к антиобщественному поведению», — к такому выводу пришли в результате обширного исследования проблемы сиротства в РФ специалисты международной правозащитной организации «Human Rights Watch». По данным Минздрава РФ, у 64 % детей, поступающих в дома ребенка, обнаруживаются осложнения внутриутробного развития, у 10 % — врожденные или наследственные заболевания. 30–40 % детей поступают из семей алкоголиков, 50 % — от матерей и 13 % — от отцов, больных туберкулезом, сифилисом, диабетом или имевших психические заболевания. Так, например, в детских домах Пермской области насчитывается менее 1 % здоровых детей. Отставание в физическом развитии наблюдается почти у половины сирот. Ненадлежащие условия содержания, невозможность обеспечить нормальный уход за больными усугубляют ситуацию. Большинство детей страдают внутриутробными инфекциями, такими, как цитомигалия, таксоплазмоз, хламидиоз. При «тяжелой олигофрении», когда ребенок признается страдающим имбецильностью или идиотией, он направляется в закрытые учреждения при Минтруда и социального развития, где пробудет до 18 лет, не получая практически никакого образования, чтобы потом перевестись во взрослое учреждение такого же типа. Часто ставящийся диагноз «легкая степень олигофрении» означает, что в процессе школьного обучения сирота будет подвергаться более жестким дисциплинарным взысканиям, чем его сверстник, которого родители оставили в детском доме только на время и навещают лишь изредка. Известны случаи, когда наказанием за шалость становилось обследование и лечение в психиатрической клинике. Как отмечает председатель Комитета защиты прав воспитанников детских домов и школ-интернатов города Москвы В. Семенов, в школе-интернате №8 направление детей в психиатрические больницы используется систематически и объясняется не медицинскими показаниями, а плохим поведением, неуспеваемостью и т. д. В больнице они получают такие сильные препараты, как аминазин, галоперидол, тизерцин, реланиум, которые могут серьезно повредить здоровью ребенка. Галоперидол, например, часто вызывает трудности мышления, снижает способность концентрироваться, притупляет эмоции, вызывает ночные кошмары, депрессию, половую дисфункцию. Гражданская комиссия по правам человека в одном из своих отчетов приводит рассказ воспитанника детского дома, отправленного в психиатрическую больницу за то, что он разбил окно, играя в футбол. Нейролептики, которыми его лечили психиатры, оказали тормозящее действие на его нервную систему: «Вернувшись из больницы, я не смог учиться, даже не мог складывать числа, и меня отправили в школу для умственно отсталых детей, хотя я уже закончил 6 классов по обычной программе. И началась моя жизнь в психоинтернатах. Как говорит главный психиатр Андроповского психоневрологического интерната, где я сейчас живу: «Сюда дорога открыта, а отсюда — только на кладбище».
Сироты с раннего детства привыкают осознавать себя «деклассированными элементами», отмечают психологи. Вырастающая из такого самоощущения предрасположенность к асоциальному образу жизни, совершению противозаконных действий и самоубийству подогревается самой атмосферой детского дома и воспитанием, которое нередко сопровождается серьезными психологическими травмами и, главное, не предусматривает приобретения навыков, способствующих «постинтернатной адаптации».
Многие дети попадают в интернатные учреждения после длительного пребывания в доме ребенка, в неблагополучной семье, на улице. Пережитые за это время стрессы сказываются на развитии эмоционально-волевой сферы. По наблюдениям психологов, для воспитанников обычных детских домов характерны односторонность, бедность поведенческих мотиваций, постоянная зависимость от поведения взрослого. В конфликтных ситуациях они зачастую неспособны сколько-нибудь объективно оценить ситуацию, не могут владеть своим настроением и поведением. Затруднения в формировании самосознания приводят к постоянному подражанию другим, поверхностности чувств, агрессивности. Пытаясь самоутвердиться, воспитанник детского дома демонстрирует свою самостоятельность отказом от подчинения правилам и моральным нормам.
Авторитарный стиль воспитания, распространенный в большинстве российских детских домов, где воспитатели в силу профессиональной беспомощности пытаются подавить ребенка физически и психологически, подвергая его жестоким и унизительным наказаниям, деформирует личность. Прямым или косвенным последствием такой деформации может стать суицид. Специалисты Республиканской клинической психиатрической больницы Удмуртии обнаружили, что в результате подобного обращения в одном из детских домов республики за 9 месяцев шесть воспитанников в возрасте от 10 до 17 лет пытались покончить с собой, один из них погиб.
Повышенная подверженность посторонним влияниям, отличающая воспитанников сиротских учреждений, часто приводит их в криминальную или полукриминальную среду. Каждый десятый из содержащихся в колониях несовершеннолетних — выходец из детского дома. Часто сироты становятся жертвами преступлений: по словам депутата Госдумы РФ Валерия Борщева, доходы от преступного бизнеса на сиротах сопоставимы лишь с доходами от торговли наркотиками. Попрошайки на улицах Москвы и других больших городов — это, как правило, сироты, которые трудятся на профессиональных мошенников, а то и воров. Причиной нарушения закона, как правило, становится полная неподготовленность воспитанников детских домов к самостоятельной жизни, которая усугубляет их и без того бедственную ситуацию: они не получают жилье или теряют его в результате обмана, не могут получить достойно оплачиваемую работу, создать полноценную семью. Проблема трудоустройства наиболее часто оказывается причиной маргинализации сирот, отмечает психолог Елена Махлах, почти 40 лет проработавшая с детьми, лишенными родительской опеки, и постоянно следящая за судьбой выпускников детских домов. Об одном из таких домов она говорит так: «Из целого выпуска десятилетней давности благополучным можно назвать лишь одного человека — он устроился в театр рабочим сцены. С остальными все очень плохо: они занялись проституцией, связались с криминалом, одна девушка убита, другая покончила с собой» (по материалам Рунета).
Понятие «человеческий материал» в применении к России в свете всего вышесказанного воспринимается, увы, почти в буквальном медицинском смысле. С таким «материалом» Россия обречена на поражение в любом серьезном противостоянии с внешним врагом и не способна справиться с врагом внутренним, как это показывают неудачи государства в борьбе с алкоголизмом и наркобизнесом в РФ. Нередко создается впечатление, что наши кремлевские мечтатели намеренно закрывают глаза на эту жизненно-важную проблему, предпочитая жить в воздушных замках своих иллюзий под песнопения околовластной челяди «Все хорошо! Все хорошо!». Это тоже своего рода наркотизация и весьма опасная в современном мире.
Сегодня, как никогда, актуален для российского государства и тех, кто им руководит, и призыв Солженицына к сбережению народа русского. Замечу, что простым физическим сбережением наш народ не спасти. Солженицын, как и Алексий II, конечно, имел в виду и духовное сбережение нашей нации, сбережение ее традиций, обычаев и ее культуры. Но это невозможно сделать без физического оздоровления нации и духовного возрождения русского народа. И конечно, необходимо возродить в русских людях прогрессивно исчезающее милосердие — слишком много в нашей стране сирот, обездоленных и ущербных людей, которые без помощи общества и государства попросту пропадают. Как никогда сегодня востребованы доброта и сострадание к нашим соплеменникам. Без этого невозможно выполнить главную задачу нынешнего поколения русских людей — сберечь русскую цивилизацию.
Данный текст является ознакомительным фрагментом.
Продолжение на ЛитРесВ наследство от папы. Десять вещей, которые достаются малышу от отца
Ученые установили, что ребенок наследует от своего биологического родителя не только внешность, но и некоторые привычки и особенности поведения.
Вам кажется, что малыш ушками и губами пошел в папу, а все остальное – от мамы? Вы ошибаетесь! Зарубежные исследователи определили целых десять совпадений отцов и детей: внешность, привычки, любимые выражения и много других характеристик на самом деле объединяют их.
Не верите? Сравните себя со своим отцом по следующим параметрам.
Ямочки на щёчках. В 90 процентах случаев милые ямочки на пухлых щечках ребенку достаются от его отца. Не верите — загляните в детский альбом своего папы.
Любовь к риску. О том, что систему поведения в критических ситуациях малыш наследует от своего отца, говорили еще древние ученные.
Уровень интеллекта. Хотите, чтобы ваш ребенок стал гениальным ученым или изобретателем – ищите ему такого же отца. Остальное сделают «правильные» гены.
Красивые волосы. Пока младенец не достигнет хотя бы возраста 1 года, в это сложно поверить. Но потом всякие сомнения развеются, когда у вашего сына появится такой же вихор на макушке, как и у его папы.
Любимая поза для сна. Все эти милые фото, на которых дети спят в совершенно одинаковых позах как и их папы – вовсе не монтаж. Это проявления закона природы. Если мужчина любит спать, широко раскинув руки в стороны, будьте уверены, что и его ребенок будет спать точно так же.
Левша или правша. Да-да, папа «в ответе» и за то, какое полушарие у ребенка будет больше развито и кем он станет – правшой или левшой.
Здоровье зубов. Если ваш муж имеет привычку раз в полгода наведываться к стоматологу «для осмотра» — это неспроста. Скорее всего, в этом виноваты его плохие зубы, а значит, такая же привычка появится вскоре и у ваших общих детей.
Чувство юмора. Вообще-то, его можно развить в любом человеке. Но именно за природное чутье смешного и курьезного отвечают папины гены.
Форма рта, подбородка, носа. Наблюдения ученых доказали, что именно в этих деталях дети чаще всего внешне похожи на отцов.
Отпечатки пальцев. Конечно, они уникальные. Но расположение линий детям передается именно от пап.
ПРАЙМ PubMed | [Врожденная гидроцефалия, олигофрения, карликовость, центростремительное ожирение и гипогонадизм; Х-сцепленное рецессивное наследственное заболевание?]
Citation
Sengers, R.C., et al. «[Врожденная гидроцефалия, олигофрения, карликовость, центростремительное ожирение и гипогонадизм; Х-сцепленное рецессивное наследственное заболевание?]». Tijdschrift Voor Kindergeneeskunde, vol. 53, нет. 1, 1985, стр. 31-4.
Sengers RC, Hamel BC, Otten BJ, et al. [Врожденная гидроцефалия, олигофрения, карликовость, центростремительное ожирение и гипогонадизм; Х-сцепленное рецессивное наследственное заболевание?]. Tijdschr Kindergeneeskd . 1985; 53 (1): 31-4.
Sengers, R.C., Hamel, B.C., Otten, B.J., van Gils, J.F., & de Pagter, A.G. (1985). [Врожденная гидроцефалия, олигофрения, карликовость, центростремительное ожирение и гипогонадизм; Х-сцепленное рецессивное наследственное заболевание?]. Tijdschrift Voor Kindergeneeskunde , 53 (1), 31-4.
Sengers RC, et al. [Врожденная гидроцефалия, олигофрения, карликовость, центростремительное ожирение и гипогонадизм; Х-сцепленное рецессивное наследственное заболевание?]. Tijdschr Kindergeneeskd. 1985; 53 (1): 31-4. PubMed PMID: 4039476.
TY — JOUR Т1 — [Врожденная гидроцефалия, олигофрения, карликовость, центростремительное ожирение и гипогонадизм; Х-сцепленное рецессивное наследственное заболевание?]. AU — Sengers, R C, AU — Hamel, B C, AU — Оттен, Би Джей, AU — Ван Гилс, Дж. Ф., AU — де Пагтер, A G, PY — 1985/2/1 / pubmed PY — 1985/2/1 / medline PY — 1985/2/1 / entrez СП — 31 EP — 4 JF — Tijdschrift voor kindergeneeskunde JO — Tijdschr Kindergeneeskd ВЛ — 53 IS — 1 N2 — Сообщается о двух пациентах мужского пола из одной семьи, которые страдают врожденной гидроцефалией, умственной отсталостью, низким ростом, ожирением и гипогенитализмом.Скорее всего, тип наследования — рецессивный, сцепленный с Х-хромосомой. SN — 0376-7442 UR — https://www.unboundmedicine.com/medline/citation/4039476/[congenital_hydrocephalus_oligophrenia_dwarfism_centripetal_obesity_and_hypogonadism L2 — http://www.diseaseinfosearch.org/result/3510 БД — ПРЕМЬЕР DP — Unbound Medicine ER —
Статьи, связанные с синдромом Алопеции, эпилепсии и олигофрении Мойнахана:
|
Семейная история задержки развития | Клиника медицины усыновления
Задержка развития у маленьких детей или умственная отсталость (ID — в прошлом называвшаяся умственной отсталостью или MR) у биологической матери или родного отца иногда указывается в справочной информации до усыновления.Это может быть указано в форме тестов IQ, специального образования или других терминов, таких как «олигофрения», которые могут означать что угодно, от истинной умственной отсталости до человека, который не закончил среднюю школу. У человека может быть более низкий IQ по множеству причин, не имеющих ничего общего с наследуемостью:
- При угрозе утопления или удушья, несчастный случай.
- Трудные роды самого родителя.
- Инфекция или менингит в младенчестве или детстве, вызвавшие повреждение головного мозга.
При этом существуют также генетические формы умственной отсталости. Однако даже генетические формы идентификации не обязательно наследуются ребенком. Существует множество режимов наследования, и некоторые из наиболее распространенных шаблонов следующие:
Доминант
Большинство наших генов состоит из пар; мы наследуем один от наших матерей и один от наших отцов. Если в одной из этих пар генов один из двух имеет изменение (или мутацию), которое само по себе вызывает умственную отсталость, это будет примером доминирующей умственной отсталости.Каждый раз, когда у нас появляется ребенок, мы случайным образом передаем по одной копии каждого гена. Следовательно, каждый ребенок, рожденный от родителя с доминантной формой ID, имеет 50:50 шанс унаследовать доминантный ген и, следовательно, иметь умственную отсталость.
рецессивный
В этом типе наследования обе копии гена должны иметь мутации, чтобы человек имел ID. Каждый раз, когда у человека появляется ребенок, он передает ему одну копию. Однако, поскольку ребенок получает вторую копию от своего другого родителя, очень маловероятно, что он или она пострадает, если только случайно другой родитель также не несет точно такой же рецессивный ген (очень маловероятно).
X-связка
Дефектный ген переносится на Х-хромосоме. У женщин две хромосомы «X», а у мужчин — одна «X» и одна «Y». Поскольку у женщин есть 2 X, один нормальный ген обычно может перекрыть дефектный. У мальчиков есть только одна копия Х-хромосомы, поэтому, если у них есть дефект, они с большей вероятностью пострадают. Хрупкий X является примером умственной отсталости, связанной с X, и у детей более старшего возраста обычно ассоциируется с некоторыми результатами физического осмотра, которые указывают на то, что ребенок может быть затронут.
Многофакторный
В большинстве семей, в которых есть история умственной отсталости, наследование более сложное, потому что оно включает несколько пар генов, а также другие факторы, которые мы также не понимаем. В этих семьях, в зависимости от того, сколько людей затронуты, вероятность того, что любой член семьи будет затронут, обычно ниже, чем при доминантном наследовании (см. Выше), но выше, чем у остальной части населения.
Чем старше ребенок, тем легче определить, может ли он иметь более низкий IQ.Если у нас есть очень маленький ребенок, рожденный от родителя (ей) с удостоверением личности, без особого семейного прошлого или анамнеза, может быть трудно сказать, какие риски для ребенка в будущем обучении. Из-за множества других факторов, которые также вызывают идентификацию, которая не является генетической (как указано выше), вероятность того, что ребенок унаследует форму генетической идентификации, все еще низка. Ключи к генетическим синдромам или формам унаследованной ID часто отмечаются при физикальном осмотре и / или на фотографиях: низко посаженные уши, широко расставленные или маленькие глаза, пороки сердца, мышечный тонус — все это должно быть проверено опытным врачом.
Еще один важный ключ к разгадке генетических форм идентификации — это родословная или количество родственников, которые также отметили проблемы развития. К сожалению, мы не часто получаем крупные родословные из других стран, но Тайвань и Корея, а также США иногда могут предоставить эту историю. Например, если ребенок — мальчик, а его брат, отец и дядя имеют удостоверения личности, то шансы, что этот ребенок также будет затронут, будет выше. Если у биологической матери задержка в развитии, но все остальные дети в семье, а также тети, дяди и двоюродные братья и сестры чувствуют себя хорошо, тогда риски будут меньше.
Сводка
- Семейный анамнез задержки развития, МР: В целом, поскольку существует так много различных причин задержки развития, которые не передаются по наследству детям, я с оптимизмом смотрю на потенциал ребенка.
- Настоятельно рекомендуется пройти медицинский осмотр и проверить фотографии у профессионала на предмет выявления синдромов или других причин задержек.
Редкие наследственные синдромы — AMBOSS
Последнее обновление: 8 марта 2021 г.
Резюме
На этой карточке представлен обзор унаследованных симптомокомплексов, которые редко встречаются в общей популяции.Эти синдромы вызваны наследственными генетическими дефектами, которые возникают либо из-за хромосомных аберраций, либо из-за аутосомных / сцепленных с полом признаков. Проявления различаются для каждого синдрома, при этом большинство особенностей связано с аномалиями развития, функциональными или структурными аномалиями различных органов. Диагноз можно подтвердить с помощью молекулярно-генетического обнаружения, флуоресцентной гибридизации in situ (FISH) или других генетических / хромосомных исследований. Лечение обычно симптоматическое.
Синдром ломкой Х-хромосомы
Хрупкой Х-хромосомы: «X-tra large» уши, яички и лицо у этих пациентов.
Синдром Прадера-Вилли и синдром Ангельмана
Обзор
- Определение: генетические синдромы, вызванные микроделецией (15q11-q13) в сочетании с геномным импринтингом.
- Этиология: возникающее в результате состояние зависит от копии пораженного гена.
- Диагноз: генетические тесты
«Прадер скучает по папе, а Ангел — по маме»: аллельная мутация / делеция отцовского происхождения при синдроме Прадера-Вилли и материнского при синдроме Ангельмана.
Синдром Прадера-Вилли
[5] [6] [7]- Клинические особенности
- Лечение
- Прогноз: нормальная продолжительность жизни, если избежать крайнего ожирения
Синдром Ангельмана
[7] [8] [9]- Клинические особенности
- Задержка умственного развития и приобретения моторики у детей грудного и раннего возраста; отсутствие речевого развития
- Умственная отсталость
- В более чем 80% случаев выраженные эпилептические припадки
- Микроцефалия
- Атаксия, тремотические движения конечностей
- Гипотония туловища, гипертонус конечностей, гиперрефлексия
- Затруднение со сном
- Характерное счастливое поведение с частым смехом (несоответствующий смех)
- Гипервозбудимость, непродолжительное внимание
- Очарование водой
- Лечение
- Прогноз: Продолжительность жизни обычно нормальная.
«Ангелы на небесах»: беспечная, легко возбудимая личность, атаксия, вербальное недоразвитие, эпилептические припадки, аномальные черты лица, тяжелая умственная отсталость.
Синдром Зеллвегера (цереброгепаторенальный синдром)
- Определение: аутосомно-рецессивный дефект гена PEX, который приводит к нарушению синтеза пероксисом [10] [11]
- Клинические особенности
- Диагноз: повышенная концентрация жирных кислот с очень длинной цепью в крови.
- Лечение
- В настоящее время не существует лекарства от синдрома Зеллвегера.
- Поддерживающее лечение, в том числе:
- Установка зонда для питания
- Консультации специалистов для лечения индивидуальных проблем (например, аудиологов, офтальмологов, ортопедов)
- Прогноз: смерть в течение одного года после рождения.
Последовательность Пьера Робена (синдром Пьера Робена)
- Определение: набор аномалий, вызывающих пороки развития ротовой полости и челюстно-лицевой области у плода [12]
- Клинические особенности
- Диагноз: флуоресцентная гибридизация in situ (FISH)
- Лечение
- При умеренной одышке: симптоматическое лечение
- Неинвазивная вентиляция
- Наблюдение и помощь во время еды
- При сильной одышке: хирургическая коррекция
- Специальные вмешательства для длительной коррекции (например,д., отвлечение нижней челюсти, чтобы язык был ближе к передней части рта)
- В случаях острого опасного для жизни респираторного дистресс-синдрома → трахеостомия
- При умеренной одышке: симптоматическое лечение
Синдром кри-дю-чат (синдром кошачьего крика)
- Определение: редкий синдром, вызванный аберрацией хромосомы 5 [14] [15]
- Эпидемиология: распределение по полу: ♀> ♂ (2: 1)
- Этиология: микроделеция короткого плеча на хромосоме 5 (46, XX, 5p- или 46, XY, 5p‑).
- Клинические особенности
- Лечение
- Симптоматическое лечение
- Ранняя психологическая и физическая помощь
- Прогноз: нормальная продолжительность жизни возможна, но зависит от сопутствующих симптомов и терапевтической помощи.
Синдром Ретта
- Определение: Х-сцепленное расстройство с прогрессирующей потерей интеллекта и когнитивных способностей, таких как речь, передвижение и мелкая моторика [16]
- Этиология: мутация Х-сцепленного доминантного гена в гене метил-CpG-связывающего белка 2 (ген MECP2)
- Обычно это не наследственный дефект гена, а спорадическая мутация.
- Мутация обычно возникает в отцовском аллеле и, таким образом, затрагивает почти исключительно женщин (при поражении плоды мужского пола умирают внутриутробно или вскоре после рождения).
- Клинические особенности
- Нормальное развитие до появления первых симптомов, что обычно происходит в возрасте 6–18 месяцев
- Симптомы регресса нервного развития, в том числе:
- Диагноз: сочетание типичных клинических проявлений и обнаружения генных мутаций.
- Прогноз: недостаточно данных относительно ожидаемой продолжительности жизни после 40 лет, так как отсутствуют долгосрочные исследования и болезнь встречается довольно редко.
Регресс Ретта: за нормальным развитием вскоре следует потеря целенаправленных движений рук и умственная и вербальная инвалидность.
Синдром Вильямса
- Определение: мультисистемное нарушение развития, вызванное делецией хромосомы 7.
- Этиология: микроделеция длинного плеча хромосомы 7 (включая делецию гена эластина).
- Клинические особенности
- Диагноз: сочетание типичных клинических проявлений и обнаружения генных мутаций.
Уильям принимает ICEcream от незнакомцев (умственные расстройства, сердечно-сосудистые пороки, эльфийские черты лица, комфорт с незнакомцами).
Синдром Смита-Лемли-Опица
Наследственная остеодистрофия Олбрайта (синдром Мартина-Олбрайта)
Синдром Нунана
Синдром Сильвера-Рассела
Синдром Тричера Коллинза
Ссылки
- Синдром Тричера Коллинза. https://www.ncbi.nlm.nih.gov/pubmed/20301704 . Обновлено: 1 января 1993 г. Доступ: 2 июля 2020 г.
- Синдром ломкой Х-хромосомы. https: // medlineplus.gov / ency / article / 001668.htm . Обновлено: 8 января 2015 г. Доступ: 7 мая 2017 г.
- Синдром ломкой Х-хромосомы. https://ghr.nlm.nih.gov/condition/fragile-x-syndrome . Обновлено: 12 декабря 2017 г. Доступ: 16 декабря 2017 г.
- Нарушения биогенеза пероксисом, спектр синдрома Зеллвегера. https://www.ncbi.nlm.nih.gov/books/NBK1448/ . Обновлено: 10 мая 2012 г.Доступ: 7 мая 2017 г.
- Карими Р., Брамфилд Т., Брамфилд Ф., Сафайян Ф., Стейн С. Синдром Зеллвегера: генетическое заболевание, которое изменяет биосинтез и метаболизм липидов. Интернет-журнал фармакологии . 2006; 5 (1).
- Страница информации о синдроме Ретта. https://www.ninds.nih.gov/Disorders/All-Disorders/Rett-Syndrome-Information-Page . . Доступ: 7 мая 2017 г.
- Mainardi PC. Синдром Кри дю Шат. Орфанет J Редкие диски . 2006; 1 (33). DOI: 10.1186 / 1750-1172-1-33. | Открыть в режиме чтения QxMD
- Шиффер РБ, Рао С.М., Фогель Б.С. Нейропсихиатрия: всеобъемлющий учебник . LWW ; 2003 г.
- Портер Ф.Д. Синдром Смита – Лемли – Опица: патогенез, диагностика и лечение. евро J Hum Genet . 2008; 16 (5): стр.535-541. DOI: 10.1038 / ejhg.2008.10. | Открыть в режиме чтения QxMD
- Синдром Смита-Лемли-Опица.
- Синдром Прадера-Вилли. https://rarediseases.org/rare-diseases/prader-willi-syndrome/ . Обновлено: 1 января 2018 г. Дата обращения: 22 июня 2020 г.
- Маскари М.Дж., Готлиб В., Роган П.К. и др. Частота однопородной дисомии при синдроме Прадера-Вилли. N Engl J Med . 1992; 326 (24): с.1599-1607. DOI: 10,1056 / nejm199206113262404. | Открыть в режиме чтения QxMD
- Синдром Прадера-Вилли. https://www.ncbi.nlm.nih.gov/books/NBK1330/ . Обновлено: 4 февраля 2016 г. Доступ: 7 мая 2017 г.
- Синдром Прадера-Вилли. https://medlineplus.gov/ency/article/001605.htm . Обновлено: 19 апреля 2016 г. Доступ: 7 мая 2017 г.
- Робб МП. Введение: Руководство по коммуникационным наукам и расстройствам, второе издание . Множественное издание ; 2013
- Синдром Ангельмана. https://www.ncbi.nlm.nih.gov/books/NBK1144/ . Обновлено: 14 мая 2015 г. Доступ: 7 мая 2017 г.
- Синдром Ангельмана. https://ghr.nlm.nih.gov/condition/angelman-syndrome . Обновлено: 28 ноября 2017 г.Доступ: 30 ноября 2017 г.
- Изолированная последовательность Пьера Робена. https://ghr.nlm.nih.gov/condition/isolated-pierre-robin-sequence . Обновлено: 2 мая 2017 г. Доступ: 7 мая 2017 г.
- Drescher F, Jotzo M, Goelz R, Meyer T, Bacher M, Poets CF. Когнитивное и психосоциальное развитие детей с последовательностью Пьера Робена. Acta Paediatr . 2008; 97 (5): с.653-656. DOI: 10.1111 / j.1651-2227.2008.00742.x. | Открыть в режиме чтения QxMD
- Бхамбани В., Мюнке М. Синдром Нунана. Ам Фам Врач . 2014; 89 (1): с.37-43.
(PDF) Наследственные нарушения обмена аминокислот у детей с умственной отсталостью
Наследственные нарушения обмена аминокислот у детей с умственной отсталостью
Ц.Энхджаргал, Д.Хишигбуян, Б.Содномцерен, П.Гантуя, Э. Б.Доржкханда,
S.Отгонжаргал
Национальный центр общественного здравоохранения, Улан-Батор, Монголия
Справочная информация: исследования показали, что 6,6% детей школьного возраста в Монголии имеют
умственную отсталость. Одной из причин заболевания может быть наследственное нарушение метаболизма амино
кислоты. Обязательная система скрининговых анализов на аминокислотные нарушения у
новорожденных внедрена в большинстве стран. Для оценки необходимости внедрения
такой системы в Монголии было проведено исследование статуса наследственных нарушений метаболизма аминокислоты
у детей с олигофренией.
Методы. Скрининговые исследования проводились на засохших пятнах крови 476 детей с диагнозом «
» с диагнозом «олигофрения», посещавших специальные школы. Уровни аминокислот в моче и крови
образцов детей с положительными результатами скрининга были определены с использованием высокопроизводительной жидкостной хроматографии
.
Результаты: скрининговый анализ выявил повышенное содержание аминокислот у двенадцати детей. Из-за
отказов опекунов четырех из двенадцати положительных детей у восьми детей было оценено состояние
аминокислот в моче и крови.Результаты оценки показали, что умственная отсталость
у двух детей не была вызвана ошибками метаболизма аминокислот. У двух
оставшихся положительных детей были определены повышенные концентрации глицина и треонина
(Gly в сыворотке 116,48 мкмоль / л, Thre 354,71 мкмоль / л и Gly в моче 337,47 мкмоль / л,
Thre 539,37 мкмоль / л у одного ребенка и сывороточный Gly 135,67 мкмоль / л, Thre 254,90 мкмоль / л и
мочевой Gly 635.27 мкмоль / л, Thre 546,23 мкмоль / л у второго ребенка), что привело к диагнозу
пиридоксин-зависимой эпилепсии. Оценка результатов анализа других четырех
детей показала гипертирозинемию (сывороточный Tyr 283,79 мкмоль / л и мочевой Tyr 94,44 мкмоль / л),
гипервалинемию (сывороточный Val 115,42 мкмоль / л и мочевой Val 439,65 мкмоль / л
), гиперлизинемия(сывороточный Lys 268,73 мкмоль / л и Lys в моче 635,27 мкмоль / л) и некетотическая гиперглицинемия
(сыворотка Gly 227.37 мкмоль / л и Gly 545,77 мкмоль / л в моче).
Заключение: Результаты исследования показывают, что одной из причин олигофрении в Монголии являются
унаследованных ошибок метаболизма аминокислот, что указывает на необходимость включения
аминокислот в программу скрининга новорожденных.
Наследственные нарушения обмена аминокислот у детей с умственной отсталостью
Международная конференция по биохимии, протеомике и биоинформатике
16-17 мая 2018 г. Сингапур
Ц. Энхджаргал, Д. Хишигбуян, Б. Содномцерен, П. Гантуя, Э-Туя, Б. Доржханд и С. Отгонджаргал
Национальный центр общественного здравоохранения, Монголия
Постеры и принятые тезисы : Biochem Anal Biochem
Аннотация :Предпосылки и цель: Опросы показали, что 6.6% детей школьного возраста в Монголии страдают умственной отсталостью. Одной из причин заболевания может быть наследственное нарушение обмена аминокислот. Скрининг новорожденных на аминокислотные нарушения является обязательным в большинстве стран. Чтобы оценить необходимость внедрения такой системы в Монголии, в этом исследовании оценивается статус наследственных нарушений метаболизма аминокислот у детей с олигофренией. Метод. Скрининговые исследования проводились на засохших пятнах крови 476 детей из спецшкол с диагнозом олигофрения.Уровни аминокислот в образцах мочи и крови детей с положительными результатами скрининга определялись с помощью системы высокоэффективной жидкостной хроматографии. Результат: скрининговый анализ выявил повышенное содержание аминокислот у двенадцати детей. Из-за отказа опекунов четырех из этих двенадцати положительных детей статус аминокислот в моче и крови был оценен у восьми детей. Результаты оценки показали, что умственная отсталость у двоих детей не была вызвана ошибками метаболизма аминокислот.У двух из оставшихся положительных детей были обнаружены повышенные концентрации глицина и треонина (сывороточный Gly-116,48 мкмоль / л, Thre354,71 мкмоль / л и мочевой Gly-337,47 мкмоль / л, Thre-539,37 мкмоль / л у одного ребенка и сыворотка Gly-135,67 мкмоль / л, Thre-254,90 мкмоль / л и Gly-635,27 мкмоль / л в моче, Thre-546,23 мкмоль / л у второго ребенка), которые приводят к диагнозу пиридоксинзависимой эпилепсии. Результаты тестов других четырех детей показали гипертирозинемию (сывороточный Tyr-283,79 мкмоль / л и мочевой Tyr-94.44 мкмоль / л), гипервалинемия (Val-115,42 мкмоль / л в сыворотке и Val-439,65 мкмоль / л в моче), гиперлизинемия (Lys268,73 мкмоль / л в сыворотке и Lys-635,27 мкмоль / л в моче) и некетотическая гиперглицинемия (сыворотка Gly-227,37 мкмоль / л и Gly-моча 545,77 мкмоль / л). Заключение. Результаты исследования показывают, что одной из причин олигофрении в Монголии являются наследственные ошибки метаболизма аминокислот, и этот факт указывает на необходимость включения аминокислот в программу скрининга новорожденных. [электронная почта защищена]
Наследственный спастический парапарез: обзор новых разработок
Наследственный спастический парапарез (HSP) или синдром Стрюмпелла-Лоррена — это название гетерогенной группы наследственных заболеваний, основной клинической особенностью которых является прогрессирующая спастичность нижних конечностей.До появления молекулярно-генетических исследований этих расстройств было предложено несколько классификаций, основанных на способе наследования, возрасте появления симптомов и наличии или отсутствии дополнительных клинических признаков. Описаны семьи с аутосомно-доминантным, аутосомно-рецессивным и X-сцепленным наследованием.
Исторические аспекты
В 1880 году Струмпель опубликовал то, что считается первым четким описанием HSP. Он сообщил о семье, в которой два брата страдали спастической параплегией.Считалось, что отец был «немного хромым», что предполагало аутосомно-доминантную передачу 1. И Струмпель, и Лоррен добавляли похожие случаи в литературу в последующие годы. 3 Вскоре после этих знаковых описаний в литературе появились многочисленные сообщения о случаях HSP. Однако Пратт, который считал наличие дополнительных неврологических признаков несовместимым с исходными описаниями, назвал многие из них синдромами HSP плюс.4 В ранних обзорах этих и других случаев была сделана попытка идентифицировать «чистые» случаи, как это было описано Струмпеллом. .Однако определение «чистого» варьировалось среди авторов5-8. Именно Хардинг в 1981 году после детальной клинической оценки 22 семей предложил критерии для классификации HSP на чистые и сложные формы, которые с тех пор были приняты и обсуждаются ниже. 9 Дальнейшее подразделение чистого HSP было также предложено Хардингом в зависимости от возраста начала заболевания. Было обнаружено, что семьи можно разделить на две группы: одна с дебютом до 35 лет (тип I), а другая с дебютом после 35 лет (тип II).Казалось, что между группами имелись клинические различия: пациенты типа I имели медленное и вариабельное течение по сравнению с более быстро развивающимся типом II, у которых мышечная слабость, симптомы мочеиспускания и потеря чувствительности были более выраженными.9 Ни одна из этих классификаций не является идеальный, со многими семьями, которые не легко соответствуют критериям. Как и в случае с другими наследственными нейродегенеративными заболеваниями, раскрытие молекулярно-генетических механизмов, лежащих в основе наследственного спастического парапареза, без сомнения, предоставит более полезную и актуальную классификацию.
Эпидемиология
Распространенность HSP варьируется в разных исследованиях. Такие различия, вероятно, связаны с сочетанием различных диагностических критериев, различных эпидемиологических методологий и географических факторов. Некоторые исследования, в которых использовались аналогичные критерии и методы, показали, что распространенность HSP / 100000 составляет 2,7 в Молизе, Италия, 4,3 в Валле-д’Аоста, Италия, и 2,0 в Португалии.10-12 В этих исследованиях использовались диагностические критерии, предложенные Хардингом. и задействовали все медицинские учреждения и различных специалистов в области здравоохранения для установления случаев заболевания в конкретном регионе.Polo и др. в 1991 г. сообщили о более высоком уровне распространенности — 9,6 на 100 000 в Кантабрии, Испания.13 В их отчете для установления случаев использовались только больничные записи, хотя многие вторичные случаи были выявлены путем обследования всех родственников из группы риска.
Клинические особенности
ЧИСТЫЙ НАСЛЕДСТВЕННЫЙ СПАСТИЧЕСКИЙ ПАРАПАРЕЗ
Критерии, которые были предложены для диагностики чистого HSP, включены в таблицу 1. Большинство пациентов испытывают затруднения при ходьбе или нарушения походки, заметные либо ими самими, либо родственниками.У тех, кто дебютирует в детстве, нередко задержка ходьбы. Другие симптомы включают жесткость ног, нарушение мочеиспускания и преждевременное ношение обуви. До 25% больных протекают бессимптомно, что подчеркивает зачастую доброкачественный характер заболевания и важность тщательной клинической оценки семей, включенных в генетические исследования. Возраст дебюта может быть от младенчества до восьмого десятилетия.9 Заметные межсемейные вариации в возрасте дебюта были одним из первых указателей на генетическую гетерогенность этого состояния.Это изменение также может быть частично связано с трудностями в установлении точной даты начала, особенно у пожилых пациентов, которые болеют этим заболеванием в течение десятилетий, или может отражать другие, еще неизвестные генетические или изменяющие окружающую среду факторы.
Таблица 1Предлагаемые диагностические критерии чисто наследственного спастического парапареза и клинические признаки, которые могут предупредить врача о возможных альтернативных диагнозах
Кардинальные отклонения при обследовании пациентов с чистым HSP включают спастичность, гиперрефлексию и подошвенные разгибатели со слабостью пирамидального распределения в нижних конечностях.Спастичность нижних конечностей является заметной находкой при осмотре, особенно в области подколенных сухожилий, четырехглавой мышцы и лодыжек. Этот образец гипертонуса отвечает за классическую походку, при которой пострадавший демонстрирует циркумдукцию и ходьбу на пальцах ног. Присутствие мышечной слабости наблюдается в подвздошно-поясничной, передней большеберцовой мышце и, в меньшей степени, в подколенных сухожилиях. Характерной чертой HSP, которая подчеркивалась несколькими авторами, является заметное несоответствие между часто выраженной спастичностью и только легкой или отсутствующей мышечной слабостью.8 14 Это демонстрирует пациент с HSP, который прикован к инвалидной коляске из-за спастичности, но при ручном мышечном тесте имеет нормальную мощность.
Другие особенности, которые некоторые авторы включили в понятие чистого HSP, включают: легкие сенсорные аномалии в нижних конечностях, отсутствие толчков в голеностопном суставе, кавусную стопу, симптомы мочеиспускания, легкую атаксию в верхних конечностях и умеренное истощение дистальных отделов мышц. 15-18 Этот более инклюзивный подход, кажется, подтверждается мультисистемным участием, предложенным параклиническими исследованиями и невропатологическими находками.Нарушение чувствительности наблюдается в 10–65% случаев чистого HSP и чаще, но не исключительно, встречается у пациентов с длительным заболеванием. Обычно она заключается в снижении чувствительности к вибрации и, реже, в ослаблении чувства положения суставов в конечностях нижних конечностей. 15 16 У большинства пациентов исследования нервной проводимости в норме. 15 19 20 Это говорит о том, что аномалии, обнаруженные при обследовании, связаны с центральной аксонопатией, а не с поражением периферических нервов. Необычное обнаружение отсутствия рефлексов голеностопного сустава у пациентов с HSP, вероятно, отражает тот же процесс.9 16 Pes cavus — не такая распространенная находка, как предполагалось изначально. Хардинг в своей серии исследований обнаружила кавусную мозоль только у одной трети пациентов.9 Ее наличие может отражать тяжесть или продолжительность заболевания. Нарушение сфинктера мочевого пузыря встречается у 50% пациентов. Обычно это проявляется в сочетании срочности, неуверенности и частоты и чаще встречается при длительном заболевании. 15-17 21 год 22 Вовлечение анального сфинктера необычно для HSP, хотя Schetlens et al действительно сообщили о семье, в которой присутствовали позывы на фекалии и недержание мочи, и Шварц ранее описал человека, страдающего аналогичным образом.В обоих отчетах также присутствовали мочевые симптомы. 24 Сексуальная дисфункция описана только в одной семье. Это произошло примерно через 10–15 лет после начала заболевания, поэтому репродуктивная способность пациента не сильно пострадала. Проблема у пораженных мужчин заключалась в эректильной недостаточности с сохранением способности к эякуляции.25 Легкое мышечное истощение хорошо известно, хотя и редко встречается при HSP. Когда он присутствует, он обнаруживается в дистальных мышцах нижних конечностей, обычно в небольших мышцах стопы и передней большеберцовой мышце.Чаще встречается у пациентов, которые болеют этим заболеванием более 10 лет. 15 Если истощение мышц заметно и присутствует на ранних стадиях заболевания, диагноз следует пересмотреть. Поражение верхних конечностей относительно редко и обычно проявляется легкой гиперрефлексией, которая может присутствовать на ранних стадиях заболевания. След терминальной дисметрии также может быть обнаружен в верхних конечностях, но более ярких признаков мозжечка не наблюдается. Иногда поступают сообщения о легкой атрофии дистальных мышц, но более выраженное поражение верхних конечностей со спастичностью, слабостью или амиотрофией обычно не наблюдается при чистом HSP.9
Важными отрицательными результатами обследования являются нормальная функция черепных нервов и отсутствие признаков поражения кортикобульбарного тракта. Возможное участие вегетативной нервной системы в HSP систематически не изучалось, хотя Картлидж и Боун, сообщая об обнаружении нарушения сфинктера у пациентов с HSP, рассматривали участие вегетативной нервной системы у трех пациентов и выполняли ряд автономных функций. тесты, которые были нормальными.17
Прогноз и тяжесть HSP варьируются между семьями и, в меньшей степени, внутри одной семьи, хотя ожидаемая продолжительность жизни нормальная.Несколько авторов поддержали наблюдение Хардинга о том, что при раннем начале HSP (<35 лет) болезнь прогрессирует медленно, большинство пациентов остаются амбулаторными на протяжении большей части своей жизни, и лишь небольшая часть приковывает к инвалидному креслу в пожилом возрасте. Это контрастирует со многими случаями позднего начала HSP (> 35 лет), при которых заболевание может прогрессировать быстро, и большинство пациентов теряют способность ходить в возрасте от 60 до 70 лет. 16 20
СЛОЖНЫЙ НАСЛЕДСТВЕННЫЙ СПАСТИЧЕСКИЙ ПАРАПАРЕЗ
В то время как в чистом HSP спастический парапарез является заметной особенностью, в сложном HSP это один из компонентов гораздо более вариабельного фенотипа.Некоторые типы сложных HSP чрезвычайно редки и, возможно, были описаны только в одной семье. В некоторых случаях можно утверждать, что более чем одно наследственное заболевание может влиять на клинические особенности. Наследственный спастический парапарез был связан со многими состояниями, включая атрофию зрительного нерва, ретинопатию, экстрапирамидное заболевание, амиотрофию, деменцию, атаксию, умственную отсталость, глухоту, итиоз, периферическую невропатию и эпилепсию. Ссылки и краткие описания дополнительных клинических признаков приведены в таблице 2.
Таблица 2Клинические особенности, которые могут проявляться помимо спастического парапареза при осложненном наследственном спастическом парапарезе
Эпилепсия, связанная с HSP, не упоминалась в более ранних обзорах, возможно, потому что, имея только редкие сообщения о случаях одного или двух затронутых членов в родословной, связь могла быть случайной, учитывая высокую распространенность эпилепсии в сообществе. Однако за последнее десятилетие несколько авторов опубликовали отчеты о случаях, в которых синдромы эпилепсии и HSP, по-видимому, тесно связаны.39 41-44 Нет четкой закономерности между семьями, в которых существует эта ассоциация. Наблюдается как аутосомно-доминантное, так и рецессивное наследование. Не существует последовательного типа приступов, связанных с HSP, и сообщалось о миоклонической, простой парциальной, сложной парциальной и генерализованной эпилепсии. У пострадавших членов одной семьи иногда описывались различные типы припадков. Точно так же существует вариативность относительно того, когда приступы начинаются, с примерами припадков до и после начала нарушения походки.В этих семьях часто описывались сопутствующие умственная отсталость или когнитивные нарушения. Эпилепсия является характерным признаком митохондриальных заболеваний (например, MERRF), группы заболеваний, которые часто проявляются различными симптомами, не похожими на сложные HSP. Как будет обсуждаться позже, доказательства участия митохондрий были обнаружены в некоторых аутосомно-рецессивных семьях HSP, и будет интересно определить, вносит ли митохондриальная дисфункция вклад в патогенез других форм HSP.
Есть семьи с HSP, у которых, по-видимому, действительно поражены периферические нервы. Шади и Смит сообщили о семье с бессимптомным сенсорным поражением, обнаруженным при клиническом обследовании. У всех членов семьи, которые проходили исследования нервной проводимости, были снижены потенциалы сенсорного действия. Трем пациентам была сделана биопсия нерва, которая показала уменьшение количества миелинизированных волокон большого и среднего размера без признаков демиелинизации, что свидетельствует об аксональной невропатии. 50 Такие семьи представляют собой примеры сложных HSP.В более ранних сообщениях описывалась тяжелая сенсорная нейропатия, связанная с HSP, с хроническими безболезненными кожными язвами и невропатической резорбцией кости, происходящей в раннем детстве.51 В менее тяжелой форме трофические изменения кожи и язвы на ногах развиваются во взрослой жизни, наложенные на более длительные спастические парапарез.52 53
Деменция была связана с HSP в родословных, у членов которых могут быть дополнительные признаки, такие как атаксия, 65 дизартрия, 66 дизартрия и легкая неподвижность лица, 40 сердечное заболевание, 32 эпилепсия, 39 и синдром Маста, который состоит из спастической параплегии с деменцией, дизартрии и т. Д. и атетоз.62 О деменции также сообщалось как в аутосомно-доминантных, так и в рецессивных семьях, как об изолированном сопровождении спастического парапареза. 35–38 В этих семьях были обнаружены сходные модели когнитивных нарушений. К ним относятся различные комбинации дефектной недавней памяти, нарушения внимания, низкой скорости восприятия, плохой зрительно-моторной координации и забывчивости. В описаниях пострадавших не было признаков, указывающих на серьезное поражение коры, таких как дисфазия, агнозия и дискалькулия.Это предполагает, что подкорковая деменция может характеризовать сложный фенотип HSP спастической параплегии с деменцией. Возможно, что более легкие степени когнитивных нарушений могут быть более частыми, чем ранее предполагалось, у людей, затронутых HSP.38 Эта проблема еще не подвергалась систематической оценке. В одном из немногих исследований, в которых тщательно оценивалась когнитивная функция, бессимптомные нарушения были продемонстрированы у пяти из семи пациентов из четырех семей с чистым HSP.67 Таким образом, кажется вероятным, что если пациентов тщательно обследовать на предмет субклинических когнитивных проблем, то даже чистый HSP окажется мультисистемным расстройством с вовлечением вне двигательной системы.Этот феномен все чаще обнаруживается при других явно селективных двигательных расстройствах, включая заболевание двигательных нейронов68. 69
Результаты расследования
Исследования полезны для исключения альтернативных диагнозов, но не повышают достоверность диагностики HSP. Несколько авторов сообщают, что время центральной моторной проводимости показывает либо не регистрируемые, либо отсроченные ответы от нижних конечностей, с обычно нормальными значениями от верхних конечностей.70-73 Сообщается, что соматосенсорные вызванные потенциалы малы или отсутствуют, при этом аномалия снова наблюдается в основном в нижних конечностях. 72 74 Исследования нервной проводимости и ЭМГ в большинстве случаев являются нормальными показателями чистого HSP.9 20 73 75 76
Анализ CSF обычно ничем не примечателен при HSP. Однако случайные сообщения об аномалиях включали повышение концентрации белка в сложных семьях и повышение концентрации гомокарнозина в сложной семье со спастической параплегией, прогрессирующей умственной недостаточностью и пигментацией сетчатки.18 29 77 78
На МРТ спинной мозг может казаться маленьким, но другие аномалии обычно не видны. Время от времени поступают сообщения о легкой или умеренной атрофии внутричерепных структур, особенно мозолистого тела, а также о поражениях белого вещества в полушариях головного мозга. 71 79 80 Однако заметная атрофия или изменения белого вещества на МРТ требуют тщательного исключения других состояний.
В молекулярной генетике HSP наблюдается быстрый прогресс, который повлияет на то, как мы будем исследовать и диагностировать HSP в будущем, и это будет обсуждаться позже.
Дифференциальный диагноз
Наследственный спастический парапарез следует рассматривать как диагноз исключения. Важно учитывать поддающиеся лечению расстройства, которые могут проявляться аналогичным образом, такие как дефицит B12, допа-зависимая дистония и структурные нарушения спинного мозга. Заболевания, прогноз которых значительно отличается от HSP, такие как рассеянный склероз и семейное заболевание двигательных нейронов, также должны быть исключены. Необходимость исследований будет зависеть от индивидуальной клинической картины.В таблице 3 подробно описаны исследования, которые могут быть полезны для исключения других расстройств у конкретных пациентов. Только после уверенного исключения других диагнозов пациенты и их семьи могут получить соответствующую консультацию.
Таблица 3Дифференциальный диагноз наследственного спастического парапареза
Невропатология
Наследственный спастический парапарез — это заболевание, совместимое с нормальной продолжительностью жизни, при этом большинство пациентов умирают в старости от случайных болезней. Учитывая хронический характер заболевания и ограниченное количество вариантов лечения, значительная часть пациентов может не проходить регулярное обследование у невролога.Поэтому неудивительно, что сообщений о патологии HSP относительно мало. Шварц и Луи проанализировали ранние патологические отчеты в 1952 году, идентифицировали семь случаев чистого HSP и добавили свое собственное описание случаев в 1956 году.81 24 С тех пор было опубликовано лишь несколько патологических отчетов80. 82-84 Основным невропатологическим признаком чистого HSP является дегенерация аксонов, которая максимальна в терминальных частях наиболее длинных нисходящих (кортикоспинальные тракты) и восходящих (пути дорсального столба) трактов спинного мозга.Демиелинизация и глиоз могут сопровождать потерю аксонов. Наиболее серьезно поражены проводящие пути — это пересеченные и непересеченные кортикоспинальные пути к нижним конечностям и волокна fasiculus gracilis от нижних конечностей (рисунок). Поражение спиноцеребеллярных трактов наблюдается примерно в 50% случаев. Другие менее распространенные результаты включают уменьшение количества клеток Беца в слое V моторной коры в четырех случаях, заметную потерю нейронов в столбце Кларка в других четырех случаях и один случай, когда мозжечок и базальные ганглии показали потерю нейронов.Ганглии задних корешков, задние корешки и периферические нервы обычно в норме, как и клетки передних рогов, хотя в одном отчете действительно продемонстрирована дегенерация клеток передних рогов по всему спинному мозгу пострадавшего человека.
Срез спинного мозга средней шейки матки из случая сцепленного с хромосомой 2 аутосомно-доминантного чистого HSP (SPG4), окрашенного на миелин Luxol Fast Blue. Имеется диффузная миелиновая бледность пересеченных и непересеченных кортикоспинальных трактов. Кроме того, изящные пучки в центре спинных колонн демонстрируют заметную миелиновую бледность.Клетки переднего рога нормальные по количеству и морфологии.
В единственном количественном исследовании было обнаружено, что размер пирамид у пострадавшего в возрасте 57 лет сопоставим с размером пирамид в среднем у двухлетнего человека. Общее количество миелинизированных нервных волокон в пирамиде составило 376 055 по сравнению с 625 700 у 20-летнего мужчины. В том же исследовании количество клеток Беца было уменьшено до 23 652 в левой моторной области по сравнению с 34 562 у 22-летней женщины. Природа клеточной дегенерации неясна.Уменьшение количества клеток Беца можно объяснить ретроградной дегенерацией, которую можно увидеть, когда повреждение спинного мозга происходит по нескольким механизмам. Учитывая, что дистальная часть длинных аксонов в кортикоспинальных трактах, а также сенсорные и спиноцеребеллярные пути преимущественно затрагиваются в HSP, было высказано предположение, что происходит процесс «отмирания назад», с дегенерацией, начинающейся дистально, а затем продолжающейся по направлению к телу клетки. 0,24
Патологические отчеты иногда выявляют клинически неожиданные проблемы, такие как дегенерация спинного отдела позвоночника у пациентов без обнаруживаемых сенсорных аномалий в течение жизни.В одном случае была задокументирована патология мозжечка и базальных ганглиев при аутопсии без каких-либо задокументированных клинических коррелятов.
Недавно были описаны новые патологические изменения в случае чистого HSP с аутосомно-доминантным наследованием, сцепленного с локусом хромосомы 2. Описанный пациент имел ранний анамнез, полностью совместимый с чистым HSP, у него развивался прогрессирующий спастический парапарез в возрасте 19 лет. Однако в возрасте 72 лет у него развилось быстро прогрессирующее дементирующее заболевание с несколькими эпилептическими припадками на претерминальных стадиях.Патология спинного мозга полностью соответствовала чистому HSP. Однако патология головного мозга была необычной. Интересные особенности включали потерю нейронов в черной субстанции с ассоциированными тельцами Леви и бледными тельцами, а также изменения гиппокампа с тау-иммунореактивными нейрофибриллярными клубками. Другой примечательной особенностью было наличие тау-положительных телец включения в лимбической области и неокортексе. Эти изменения ранее не наблюдались при HSP и также не соответствуют патологии, характерной для других определенных форм деменции.Поэтому возможно, что эти церебральные патологические особенности являются прямым результатом экспрессии аномального продукта гена SPG440.
Генетика
Клиническая гетерогенность HSP сочетается с генетической гетерогенностью. Аутосомно-доминантные, рецессивные и X-сцепленные семьи хорошо распознаются с несколькими описанными локусами. Краткое изложение текущей генетической классификации HSP показано в таблице 4.
Таблица 4Современная генетическая классификация наследственного спастического парапареза
Аутосомно-доминантный HSP (ADHSP)
Аутосомно-доминантное наследование является наиболее распространенным типом наследования при HSP и связано с локусами на хромосоме 2p (SPG4), 38-40 71 86-94 хромосома 14q (SPG3), 88-90 95-97 хромосома 15q (SPG6), 98 хромосома 8q (SPG8), 99 хромосома 10q (SPG9), 64 и хромосома 12q (SPG10).100
ХРОМОСОМ 2P LOCUS SPG4
Локус хромосомы 2 (SPG4) на сегодняшний день является наиболее распространенным локусом, с которым связаны семейства ADHSP, составляя около 40% родословных. Недавно ген, ответственный за HSP в локусе SPG4, был идентифицирован с использованием стратегии позиционного клонирования. Ген спастин расположен на хромосоме 2p22-p21 и состоит из 17 экзонов, охватывающих область размером 90 т.п.н. 101. Спастин является членом группы белков, известных как АТФазы, связанные с различными клеточными активностями (AAA).Белки ААА участвуют в различных клеточных функциях, включая регуляцию клеточного цикла, деградацию белков, биогенез органелл и функцию белка, опосредованную пузырьками.102 Все эти клеточные механизмы включают сборку и функцию белковых комплексов, и было высказано предположение, что действуют члены семейства ААА. как белки-шапероны в этих комплексах.103 Белки AAA имеют общий домен из 230 аминокислот, который обеспечивает функцию АТФазы. Вне этого домена наблюдается очень небольшая гомология, за исключением близкородственных белков того же подсемейства белков AAA.102 Интересно, что параплегин, член другой подгруппы белков AAA, как было показано, участвует в аутосомно-рецессивном HSP SPG7 (см. Ниже) .104 Спастин имеет небольшую гомологию с параплегином, за исключением домена AAA и, в отличие от параплегина, предположительно имеет ядерная локализация. Спастин демонстрирует большую гомологию с конкретным подклассом белков ААА, которые включают субъединицы 26S протеасомы дрожжей.101 Было высказано предположение, что ядерные белки ААА, которые проявляют гомологию с субъединицами 26S протеасомы, играют косвенную роль в регуляции генов, вызывая протеолитическую активацию или деградацию факторов транскрипции. .105
На сегодняшний день идентифицированные мутации спастина включают бессмысленные, бессмысленные и сплайсинговые мутации в различных экзонах, приводящие к серьезным изменениям в аминокислотной последовательности домена AAA. Это предполагает, что SPG4 вызывается потерей функции, что означает, что пороговый уровень экспрессии белка спастина имеет решающее значение для сохранения аксонов в кортикоспинальном тракте.101
Хотя большинство родословных SPG4 имеют чистый HSP, было три семейства со сложными фенотипами, также связанными с этим локусом.38-40 В чистых семьях HSP, связанных с этим локусом, наблюдаются выраженные межсемейные и внутрисемейные вариации по степени тяжести и наличию или отсутствию таких особенностей, как симптомы со стороны мочевого пузыря и снижение чувствительности к вибрации.
Не все опубликованные исследования комментируют серьезность двигательной дисфункции у пострадавших людей, а у тех, кто часто это делает, непросто напрямую сравнивать различные отчеты. Существует тенденция к разной степени тяжести, при которой большинство пострадавших людей сохраняют способность ходить самостоятельно или с минимальной поддержкой.В крайнем случае — бессимптомные пациенты с пирамидными признаками в нижних конечностях с нормальной или лишь слегка ненормальной походкой и несколько пациентов, прикованных к креслу или прикованных к постели71. 94
Существует заметная вариабельность возраста начала симптомов в семьях, сцепленных с хромосомой 2p. Несколько опубликованных родословных показывают возраст появления симптомов в пределах 5 или более десятилетий у разных членов одной семьи. Средний возраст появления симптомов из опубликованных родословных колеблется от 12 до 43 лет, что также свидетельствует о межсемейных вариациях.Причина такой межсемейной и внутрисемейной изменчивости неизвестна. Он может представлять аллельную гетерогенность или присутствие модифицирующих генов или факторов окружающей среды. Другим фактором может быть очевидная сложность определения точного времени появления симптомов, которые начинаются незаметно и часто присутствуют в течение многих лет.
В некоторых семьях сообщается, что в последующих поколениях возраст начала заболевания снижается40. 71 89 92 Это наблюдается при других нейродегенеративных заболеваниях, например, болезни Хантингтона, зубочелюстно-паллидолизийской атрофии (DRPLA) и болезни Мачадо-Джозефа (MJD).Однако эти заболевания вызваны аномальной экспансией повторов CAG, и именно увеличение длины экспансии повторов коррелирует с более ранним началом заболевания в последующих поколениях.106-108 До открытия гена спастина велись обширные поиски доказательства того, что SPG4 был нарушением тринуклеотидных повторов. Как клинические, так и молекулярно-генетические доказательства того, являются ли расширенные повторы CAG патогенными в SPG4, неубедительны.109-111 Однако ген спастина не содержит аномально увеличенного повтора CAG, и должно быть альтернативное объяснение очевидного ожидания, наблюдаемого в некоторых семьях SPG4. искать.Это может быть просто так, когда сообщается об ожидании, это может фактически отражать предвзятость установления, при которой заболевание выявляется в раннем возрасте у детей пострадавших родителей.
Интересно, что в трех семьях, сцепленных с хромосомой 2p, HSP был связан с когнитивными нарушениями. Уэбб и др. описали большую семью с 12 больными членами. Один пациент умер от слабоумного заболевания в возрасте 62 лет, а у четырех членов семьи с HSP при нейропсихологическом тестировании было обнаружено слабоумие подкоркового типа.38 Во французской семье, описанной Heinzleff et al , четверо из семи затронутых членов семьи имели когнитивные нарушения, тогда как один имел тяжелую деменцию с некоторыми корковыми особенностями. Трое затронутых членов с когнитивными нарушениями также страдали эпилепсией.39 В семье, о которой сообщили White et al , член индекса с HSP умер с поздним началом тяжелого дементирующего заболевания с корковыми и экстрапирамидными особенностями. Два других члена семьи имели нарушение памяти в пожилом возрасте, хотя подробная информация о двигательных симптомах у этих членов отсутствовала.Еще один член семьи с HSP имел пограничные трудности в обучении с легкими нарушениями всех аспектов памяти. Как и в других семьях, были члены семьи с HSP без когнитивных изменений. Патологические находки у пациента с деменцией в этой родословной обсуждались ранее40.
ХРОМОСОМ 14Q LOCUS SPG3
Первым обнаруженным локусом ADHSP был SPG 3 на хромосоме 14q, и только пять опубликованных семейств были сцеплены с этим локусом. Расположение гена SPG3 на хромосоме 14q11.2-q24,3 сузился до области 7 см. 88 90 95–97 Среди пяти семей действительно наблюдается тенденция к относительно раннему возрасту начала заболевания, при этом среднее значение обычно находится в пределах первого или второго десятилетия. Не было никаких доказательств того, что предвосхищение возраста наступает в следующих поколениях. Первоначально считалось, что фенотип SPG 3 также может быть более серьезным, чем другие подтипы HSP. Однако Хуанг и др. и Гисперт и др. опубликовали родословные, в которых доля серьезно пораженных больных (нуждающихся в инвалидной коляске) не отличается от той, которая наблюдается в некоторых родословных, связанных с SPG4.
По сравнению с другими локусами, о которых уже упоминалось, в фенотипе SPG 3, по-видимому, отсутствует участие мочевого пузыря или органов чувств.
ХРОМОСОМА 15Q LOCUS SPG6
Только одно большое семейство из Северной Америки было связано с SPG6. Локус был сопоставлен с интервалом ~ 7,3 сМ в центромерной области хромосомы 15q.98 Средний возраст начала заболевания составлял 22 года (12–35), и в целом фенотип был чистым HSP, хотя фенотип относительно тяжелый. .Почти треть пострадавших членов нуждалась в инвалидной коляске, некоторым из них было уже 40 лет. Pes cavus почти всегда присутствовал у пораженных членов, что, возможно, отражает тяжесть заболевания.
ХРОМОСОМА 8Q ЛОКУС (SPG8)
Две семьи были сцеплены с локусом SPG8 на хромосоме 8q24. Одна большая североамериканская семья, а в последнее время и британская семья, позволили уточнить генетический локус до области 3,4 сМ.99 112 Средний возраст появления симптомов в родословных, связанных с SPG8, был сопоставим с семьями SPG4.Фенотип казался довольно серьезным, особенно в американской семье, в которой 10 из 15 пораженных членов старше 40 лет требовали использования инвалидной коляски, восемь из них более 50% времени.113 В обеих родословных клинические особенности были типичными для чистого HSP с некоторыми членами семьи, имеющими пониженное чувство вибрации, симптомы со стороны мочевого пузыря и кавусную мозоль. Магнитно-резонансная томография показала выраженную атрофию спинного мозга у одного пациента с умеренными симптомами из американской семьи, связанной с хромосомой 8q.Было обнаружено уменьшение площади поперечного сечения на 50% на уровне Т9 по сравнению с контрольной группой того же возраста и пола113.
Доказательства митохондриальной дисфункции как механизма заболевания в этом локусе не были обнаружены при гистохимическом или биохимическом анализе мышечной биопсии, взятой у одного пораженного члена113.
ХРОМОСОМ 10Q LOCUS SPG9
Одна крупная итальянская родословная была связана с локусом SPG9 на хромосоме 10q23.3–24.2 в регионе, охватывающем 12 сМ.64 Средний возраст появления симптомов варьируется от первого до третьего десятилетия жизни, и авторы предлагают доказательства для ожидания на протяжении трех поколений.Клинический фенотип — один из сложных HSP со спастическим парапарезом, связанным с двусторонней катарактой, гастроэзофагеальным рефлюксом с постоянной рвотой и дистальной амиотрофией, вторичной по отношению к явной аксональной моторной невропатии. Родословная со схожим сложным фенотипом врожденной катаракты и спастического парапареза была описана ранее, что также может быть связано с тем же геном в локусе SPG9114.
ХРОМОСОМА 12Q LOCUS SPG10
Последним обнаруженным локусом ADHSP является SPG10 на хромосоме 12q13, охватывающий 9.2 сМ в британской семье100. Средний возраст появления симптомов у пораженных членов семьи был моложе, чем в родословных SPG4, и составлял 10,8 (SD 9,6) лет. Фенотип был одним из чистых HSP с широким диапазоном тяжести заболевания: некоторым пациентам требовалась инвалидная коляска, тогда как другие оставались бессимптомными в возрасте 40 лет.
Reid et al описали еще одну родословную, в которой было исключено сцепление со всеми пятью известными локусами ADHSP, что предполагает присутствие по крайней мере еще одного, еще не открытого локуса ADHSP.100
Аутосомно-рецессивный HSP (ARHSP)
Рецессивное заболевание встречается гораздо реже, чем доминирующая форма. Видны как чистые, так и сложные фенотипы. Есть кровные семьи, связанные с тремя локусами: SPG5 на хромосоме 8p, 115SPG7 на хромосоме 16q116 и недавно обнаруженный локус на хромосоме 15q, 117, который в настоящее время не имеет официального обозначения в базе данных генома.
ХРОМОСОМ 8P LOCUS SPG5
Локус SPG5 находится на хромосоме 8p12-q13 и охватывает интервал 32 см.Четыре тунисские семьи, связанные с SPG5, имеют чистый фенотип со средним возрастом начала заболевания от 1 до 20 лет. Кажется, что нет разницы между клиническими характеристиками чистого ADHSP и чистого ARHSP.115
ХРОМОСОМА 16Q LOCUS SPG7
Локус SPG7 находится на хромосоме 16q24.3, и в настоящее время с этим локусом сцеплены три семейства. Фенотип неоднороден, встречаются как чистые, так и сложные семейства. Первоначальное семейство, связанное с SPG7, было описано как чистое, хотя были дополнительные признаки, обычно не наблюдаемые в чистом HSP, присутствующие в этом семействе, о которых сообщали DeMichele et al , которые включали дизартрию.116
Белок, кодируемый SPG7, получил название параплегин. Недавно в этом исходном семействе, сцепленном с SPG7, была описана гомозиготная делеция 9,5 т.п.н. в последних пяти экзонах гена параплегина на хромосоме 16q. Когда другие неродственные пациенты с HSP были подвергнуты скринингу с использованием анализа одноцепочечного подтверждающего полиморфизма (SSCP), были выявлены две дополнительные мутации сдвига рамки считывания в гене параплегина. Один произошел в семье чистых ARHSP из Италии, у пораженных членов обнаружена делеция 2 п.н. в экзоне 6.Другой был обнаружен в сложной семье ARHSP, дополнительные клинические признаки которой включали атрофию зрительного нерва с атрофией коры головного мозга и мозжечка, видимой на изображениях. Эта мутация состояла из вставки в кодирующую область последнего экзона. Все три мутации SPG7 приводят к серьезным нарушениям белка параплегина. Параплегин является членом митохондриальных металлопротеиназ, которые являются подгруппой семейства белков AAA. Другие члены этой подгруппы включают митохондриальные белки дрожжей Afg3p и Rcalp, которые проявляют как протеолитическую, так и шапероноподобную активность и локализуются на внутренней мембране митохондрий.Исследования нацеливания и локализации показали, что белок параплегина локализуется в субклеточном компартменте митохондрий. Обнаружение доказательств митохондриальной дисфункции в мышечной ткани при биопсии у затронутых членов этих семей подтверждает участие митохондрий в процессе болезни. Мышечные аномалии заключались в наличии волокон, отрицательных по цитохромоксидазе, и рваных красных волокон, которые демонстрировали интенсивное окрашивание на сукцинатдегидрогеназу, изменения, которые предполагают нарушение митохондриального OXPHOS.104 Биопсия мышцы пациента с ARHSP, сцепленным с хромосомой 8q, не выявила доказательств митохондриальной дисфункции, что позволяет предположить альтернативный механизм заболевания при этом типе HSP113.
Митохондриальная дисфункция была выявлена при некоторых нейродегенеративных заболеваниях. При таких заболеваниях, как болезнь Паркинсона, может быть прямое участие комплексов дыхательной цепи. При атаксии Фридрейха и болезни Хантингтона дефекты OXPHOS кажутся вторичными по отношению к изменениям белков, отличных от ферментов дыхательной цепи.118
Недавно ген AFG3L2, связанный с параплегином, был идентифицирован на хромосоме 18p11.119 Предсказанный генный продукт имеет гомологию с параплегином, митохондриальными АТФазами дрожжей, Afg3p и Rcalp и родственным геном AFG3L1119. 120 AFG3L2 также локализуется в субклеточном компартменте митохондрий. Требуются дальнейшие исследования, чтобы установить, насколько распространены дефекты параплегина в различных формах HSP, и установить, участвует ли AFG3L1 или AFG3L2 в возникновении HSP, особенно в семьях со сложным фенотипом, где мультисистемное вовлечение может напоминать клинические признаки, обнаруженные другие митохондриальные нарушения.
Этот класс гена может быть вовлечен в другие неврологические заболевания, и интересно, что AFG3L2 находится в области хромосомы 18p, с которой связана форма идиопатической торсионной дистонии121.
ХРОМОСОМА 15Q
После открытия SPG5 и SPG7 все еще оставались семейства ARHSP, не связанные ни с какими локусами. Murrilo и др. недавно обнаружили, что в настоящее время является наиболее распространенным локусом для ARHSP на хромосоме 15q13–15.117. Они проверили восемь семей и обнаружили доказательства сцепления с этим новым локусом в семи.
Фенотип в этих семьях варьирует, описываются как чистые, так и сложные формы. В чистых семьях возраст начала заболевания приходился на первое десятилетие в одной и в возрасте 43 лет в другой, в которой был затронут только один член семьи. Между сложными семьями также наблюдались фенотипические различия, хотя некоторые сходные черты присутствовали в более чем одной семье. Наиболее интересной была атрофия или агенезия мозолистого тела, продемонстрированная на МРТ, наблюдаемая как у пораженных братьев и сестер из одной семьи, так и у одного пораженного члена из другой.У этого последнего члена также были обнаружены перивентрикулярные изменения белого вещества. Обе эти семьи имели одинаковый возраст появления симптомов болезни во втором десятилетии жизни, и умственная отсталость была характерной чертой, отмеченной у одного члена от каждой семьи. Другими ассоциациями со спастической параплегией в этом локусе были смешанная моторная и сенсорная нейропатия в одной семье и легкая дисфункция мозжечка в двух других.
Ассоциация HSP, связанного с хромосомой 15q, с агенезом мозолистого тела и умственной отсталостью представляет интерес, поскольку было показано, что ген синдрома Андермана или агенезии мозолистого тела и периферической невропатии (ACCPN) также связан с хромосомой 15q.122 Возможно, эта форма HSP и синдром Андермана являются аллельными нарушениями с разными мутациями в одном и том же гене, приводящими к различным фенотипам.117 Это наблюдается в X-сцепленном HSP, где разные мутации в гене PLP приводят к возникновению HSP фенотип или болезнь Пелицея-Мерцбахера.
Х-сцепленный HSP
Хотя Х-сцепленный HSP встречается редко, его молекулярно-генетическая основа относительно хорошо изучена.
Существует два разных локуса, идентифицированных для X-сцепленного HSP, SPG1 и SPG2.60 123 Семьи со сложным HSP были связаны как с SPG1, так и с SPG2, тогда как чистый фенотип был связан только с SPG2.
САУ X-LINKED LOCUS
Осложненный HSP, связанный с SPG2, состоит из основного фенотипа спастического парапареза, мозжечкового синдрома и умственной отсталости. Видны межсемейные вариации, при этом основные особенности проявляются в различной степени с дополнительными признаками или без них, такими как атрофия зрительного нерва. Аналогичным образом происходит внутрисемейная изменчивость124. 125 Об этом сообщалось в более ранних описаниях X-сцепленных семей.126
Ген в локусе SPG2 представляет собой ген протеолипидного белка (PLP). Известно, что мутации, чаще всего дупликации, в этом гене вызывают болезнь Пелицея-Мерцбахера (ПМД). Это дисмиелинизирующее заболевание, поражающее ЦНС, которое состоит из ранних мозжечковых и пирамидных признаков с быстрым прогрессированием, приводящим к смерти, как правило, в младенчестве или детстве. Было показано, что сложные родословные HSP, сцепленные с локусом SPG2, имеют мутации в гене PLP в экзонах 3b, 4 или 6.127 128 Следовательно, разные мутации в одном и том же гене могут вызывать либо фенотип PMD, либо сложный фенотип HSP. PLP является основным белком миелина, участвующим как в созревании миелина, так и в поддержании его в ЦНС. Менее тяжелый фенотип и более позднее развитие признаков у SPG2 по сравнению с PMD связано с тем, что PLP имеет меньшую изоформу, DM-20. И в SPG2, и в PMD концентрации PLP снижены, тогда как концентрация DM-20 значительно снижена только в PMD. Считается, что эти два изомера немного по-разному влияют на миелин.Изоформа DM-20, по-видимому, преимущественно участвует в созревании миелина, тогда как изоформа PLP, которая экспрессируется на более поздней стадии развития, играет роль в уплотнении и поддержании миелинового листа. Миелиновые оболочки, в которых отсутствует белок PLP, будут собираться, и хотя они могут функционировать, они в конечном итоге претерпевают ускоренный оборот.129 Эти различия могут объяснять заметные фенотипические вариации между SPG2 и PMD.
Было обнаружено еще одно сложное семейство, связанное с областью, содержащей локус SPG2, без мутации, идентифицированной в гене PLP.Авторы предполагают, что это может свидетельствовать о близлежащем третьем локусе для X-сцепленного HSP.130 Однако у части пациентов с ПМД мутации не выявлены.131 132 Это может быть связано с наличием нераспознанных мутаций в некодирующих областях гена или с мутацией в соседнем гене, участвующем либо в регуляции гена PLP, либо в образовании миелина.
Чистый X-связанный HSP очень редок, на сегодняшний день в литературе описано только пять семейств.123 133-136 Два из этих семейств были сцеплены с локусом SPG2.123 133 Только в одном из этих семейств была обнаружена замена единственного основания в экзоне 5 гена PLP, показывая, что родословная чистого HSP является аллельной по отношению к сложному HSP (SPG2) и PMD.
X-LINKED LOCUS SPG1
Семьи с осложненными БТШАМИ связана с SPG1 пометили фенотипические различия по сравнению с предыдущими родословными, в числе которых отсутствие мозжечка участия, тяжелой умственной отсталость, а также врожденные аномалии опорно-двигательный аппарат, в первую очередь отсутствие разгибателей сгибателя.60 Этот фенотип обусловлен мутацией в экзоне 26 гена молекулы адгезии клеток L1 (L1CAM) 61. L1CAM представляет собой трансмембранный гликопротеин, который в основном экспрессируется нейронами и шванновскими клетками. Он играет важную роль в развитии нервной системы, участвуя в росте аксонов и поиске пути. Различные мутации в этом гене вызывают синдромы MASA (умственная отсталость, афазия, шаркающая походка и приведение больших пальцев), Х-сцепленная гидроцефалия и Х-сцепленная агенезия мозолистого тела.В семьях с мутациями в гене L1CAM существует большая межсемейная и внутрисемейная изменчивость. В одной семье могут быть члены с различными фенотипами. Заболевания теперь называют частью клинического синдрома CRASH (гипоплазия мозолистого тела, отсталость, приведение больших пальцев, спастическая параплегия и гидроцефалия) .137
Заключение
Неудивительно, что на раннем этапе возникли трудности при попытке классифицировать HSP, который представляет собой гетерогенную группу заболеваний, которые в лучшем случае могут иметь общий конечный путь в дегенеративном процессе нейронов.Молекулярно-генетическая классификация сейчас начинает определять пределы условий и их корреляции генотип / фенотип.
В настоящее время мутации в четырех генах; L1CAM, PLP, параплегин и спастин были идентифицированы как играющие роль в развитии фенотипа HSP, при этом идентичность генов в других семи локусах все еще неизвестна. Недавнее открытие генов спастина и параплегина открывает новую эру захватывающих возможностей, в которой мы можем приступить к исследованию физиологии двигательной системы и дегенеративного процесса, которому она подвергается, и, надеюсь, открывают возможности для лечения.В ближайшем будущем можно ожидать открытия остальных генов и их продуктов, что поможет нам лучше понять эти заболевания.
Наличие такой генетической гетерогенности, лежащей в основе HSP, в некоторой степени объясняет заметные фенотипические вариации, наблюдаемые между семьями. Однако обнаружение сходных внутрисемейных вариаций можно объяснить только наличием генетических модифицирующих факторов или неизвестным взаимодействием с окружающей средой. Meyer и др. продемонстрировали мутации в гене транспортера глутамата EAAT2 у двух из семи членов семьи, пораженных чистым HSP.Они постулируют, что взаимодействие с аллельным вариантом EAAT2 и неизвестным первичным генетическим дефектом может быть причиной фенотипической изменчивости, наблюдаемой в их семье138.
Несмотря на прогресс в генетике HSP, до сих пор его диагностическая значимость была относительно небольшой. Теперь это может измениться с открытием гена спастина, мутации в котором могут составлять до 50% ADHSP. При осмотре пациента с возможным HSP первым шагом остается исключить другие диагнозы.Генетический анализ даст возможность поставить точный диагноз в случаях выявления мутаций. В конечном итоге это даст возможность предсимптомной или даже пренатальной диагностики в семьях, которые хотят пройти такое обследование. Однако до тех пор, пока не будет понятен полный диапазон мутаций этих генов, техническая возможность проведения такого анализа остается неясной. Это мультиэксонные гены с различными мутациями, поэтому идентификация мутации может быть не очень простой.Тем не менее, возможность точного диагноза знаменует новую область лечения в HSP. В ближайшие годы можно ожидать дальнейшего быстрого прогресса как в идентификации новых генов, связанных с HSP, так и в клеточной биологии, лежащей в основе дегенерации двигательной системы.
